
Applying bioinformatics for antibody epitope prediction using affinity-selected mimotopes – relevance for vaccine design
- Review
- Open Access
Applying bioinformatics for antibody epitope prediction using affinity-selected mimotopes – relevance for vaccine design
- Galina F Denisova1Email author,
- Dimitri A Denisov1 and
- Jonathan L Bramson1
https://doi.org/10.1186/1745-7580-6-S2-S6
© Denisova et al; licensee BioMed Central Ltd. 2010
- Published: 3 November 2010
Abstract
To properly characterize protective polyclonal antibody responses, it is necessary to examine epitope specificity. Most antibody epitopes are conformational in nature and, thus, cannot be identified using synthetic linear peptides. Cyclic peptides can function as mimetics of conformational epitopes (termed mimotopes), thereby providing targets, which can be selected by immunoaffinity purification. However, the management of large collections of random cyclic peptides is cumbersome. Filamentous bacteriophage provides a useful scaffold for the expression of random peptides (termed phage display) facilitating both the production and manipulation of complex peptide libraries. Immunoaffinity selection of phage displaying random cyclic peptides is an effective strategy for isolating mimotopes with specificity for a given antiserum. Further epitope prediction based on mimotope sequence is not trivial since mimotopes generally display only small homologies with the target protein. Large numbers of unique mimotopes are required to provide sufficient sequence coverage to elucidate the target epitope. We have developed a method based on pattern recognition theory to deal with the complexity of large collections of conformational mimotopes. The analysis consists of two phases: 1) The learning phase where a large collection of epitope-specific mimotopes is analyzed to identify epitope specific “signs” and 2) The identification phase where immunoaffinity-selected mimotopes are interrogated for the presence of the epitope specific “signs” and assigned to specific epitopes. We are currently using computational methods to define epitope “signs” without the need for prior knowledge of specific mimotopes. This technology provides an important tool for characterizing the breadth of antibody specificities within polyclonal antisera.
Keywords
- West Nile Virus
- Lyme Disease
- Phage Display
- Conformational Epitope
- Amino Acid Pair
Introduction
Antibodies play a central role in immune memory and long-term protective responses. Serum antibodies for specific pathogens are recognized as a primary read-out for vaccination and recent studies have revealed that pathogen-specific antibodies persist for decades following vaccination [1]. It is important, however, to appreciate that not all antibodies can prevent infection. As an example, a collection of monoclonal antibodies have been isolated against the West Nile virus envelope protein (E) which recognize distinct epitopes within the protein. However, only antibodies that bind to specific epitopes can produce virus neutralization and protective immunity in vivo [2]. It was shown also that although many anti-HER2 antibodies inhibit the growth of cancer cells, some of them have no effect on cell growth, while others actively stimulate cancer growth. It has been proposed that this wide spectrum of biological effects is related to the epitope specificity of the Abs and to consequent changes in receptor signalling [3][4]. Therefore, to properly characterize a protective humoral response and to use it for vaccine design, it is necessary to characterize the epitope specificity, in addition to antibody titers.
A typical strategy for monitoring specific antibodies to known antigens involves the use of ELISAs coated with recombinant protein(s) or the vaccine itself. When combined with serologic analysis of recombinant cDNA expression libraries (SEREX), ELISAs are a powerful tool for monitoring humoral responses in various disease states where the antigens may not be known a priori [5, 6, 7]. With regard to autoimmune antibodies, the availablity of whole transcriptome sequences and effective protein expression systems has made it possible to screen the complete human proteome. Protein arrays can be used for monitoring responses to a broad range of proteins. Such arrays have been produced using genomic information [6, 7] or using information available in the SEREX database [8, 9], which collects data for antigenic proteins identified using polyserum from cancer patients. All of these methods are useful tools for measuring the magnitude and breadth of the humoral response, but they reveal little information regarding epitope specificity. While it is technically feasible to engineer recombinant proteins with specific mutations designed to disrupt putative epitopes, due to the complexity of protein folding, epitopes can be disrupted at sites distal to the mutations that will complicate interpretation of the results.
Epitope-specificity can be determined for linear epitopes by screening libraries of short synthetic peptides that span the entire target antigen (PEPSCAN) [10, 11]. However, the majority of antibody responses [12] are directed at structural epitopes which are difficult to recapitulate with synthetic peptides because they are typically formed by protein folding and, thus, are composed of amino acid residues which are often separated by great distances within the linear protein sequence. Immunoaffinity selection of random peptides offers an alternate strategy to characterize antibody epitopes because the affinity selection will identify peptides with spatially-proximal residues that may be distant from each other according to linear sequence. Indeed, this strategy offers an unbiased method to screen for epitope mimetics (mimotopes) [13] that can define antibody targets and serve directly as immunogens. To effectively apply this strategy, it is necessary to isolate many random peptides with unique sequences because each immuno-selected peptide will carry only partial homology for the original target. Through the use of computational modeling, it is possible to derive a consensus sequence from the selected peptides and identify the target epitope. The use of linear random peptides for this strategy is limited because the conformation space available to linear peptides is great, allowing the linear peptides to assume a large array of conformations. Constraining the peptide by cyclization reduces the field of conformational possibilities for the molecule and results in the peptide adopting the most favourable conformation [14]. Further, the constrained nature of these peptides causes them to adopt tertiary structure enabling them to mimic conformational epitopes. In this way, cyclized peptides present a more uniform structure than linear peptides and, thus, are preferred ligands for mapping conformational epitopes. Cyclization can be achieved simply by the incorporation of Cys residues at the N- and C-termini of a given peptide. However, production and manipulation of synthetic random peptide libraries is costly and extremely laborious, necessitating the use of biological strategies to make it feasible.
Genetic approaches for the production and manipulation of random peptide libraries
To facilitate the generation of random peptide libraries, genetic methods have been developed to introduce random peptides into scaffold proteins which can then be subjected to immunoaffinity selection followed by sequencing of the coding sequence of the random peptide. Common genetic methods for displaying random peptide ligands including: phage display, bacterium and yeast display, ribosome display and mRNA display. Incorporation of random peptides into scaffold proteins expressed on the surface of microbes (ex. phage display) are limited by the complexity of the sequence libraries that can be generated; for example, phage display libraries and bacterial display libraries are typically limited by transfection efficiency to approximately 109 independent members [15, 16, 17]. The yeast, Saccharomyces cerevisiae, is very useful as a host cell in genetic engineering because it folds and glycosylates heterologous eukaryotic proteins [18] and can be used for surface display of eukaryotic proteins and peptides in a natural conformation [19]; although, this property is not advantageous for surface display of random cyclized peptides. More recently, selection schemes based on the display of the nascent peptide chain on the surface of the ribosome have been developed [20, 21, 22]. This approach has the advantage of being fully in vitro and potentially allowing larger libraries (1012) to be explored; however, selections must be performed under conditions that preserve the integrity of the ribosome:mRNA:peptide ternary complex. The ability to synthesize covalent mRNA-peptide fusions by in vitro translation provides a different approach to the in vitro selection and directed evolution of peptides and proteins. This approach should have significant advantages over all approaches that require an in vivo step, because libraries of much greater complexity can be generated in vitro. This is a critical advantage for experiments in which a rare functional sequence is being selected from a completely random sequence initial library. Current methodologies can yield mRNA-peptide fusion libraries consisting of 1012–1013 independent members [23, 24]. However, the peptides expressed in vitrowill not be reduced and, thus, cannot be used to generate cyclized ligands.
Our work has focused on the use of phage display for the presentation of random cyclic peptides. We employ the non-lytic phage, fd, that buds from their gram-negative E. coli hosts through the periplasm where disulfide bonds are formed due to the presence of the thiol-disulfide oxidoreductase (TDOR) family of enzymes [25]. By incorporating sequences encoding random peptides with only 2 Cys residues in frame with the N-terminus of the phage pVIII coat protein, we have produced libraries of cyclized random peptide with loops ranging from 4 to 12 residues in length. Affinity-selection of phage displaying random cyclized peptides using specific monoclonal, or polyclonal, serum can yield mimetics of conformational and discontinuous antibody epitopes [26, 27] as well as carbohydrate epitopes [28, 29]. Computational modeling of the sequences of the immunoaffinity-selected mimotopes has been used with great success for the elucidation of target epitopes of monoclonal antibodies. Improved transformation methods has enabled the production of highly complex libraries [30] making phage a desirable scaffold for the manipulation of highly-diverse libraries of cyclized peptides. Affinity-selection of specific mimotopes is typically accomplished by incubating the random peptide phage library with antibodies that have been immobilized on a solid matrix. Iterative washing and binding steps allow for enrichment of phage carrying peptide inserts that are specific for the immobilized antibodies. The selected peptide sequences are then analyzed and assigned a location on the target protein using algorithms that we designed based on specific correlation analysis [31, 32] or a variety of other algorithms that have been developed based on similar principles [33, 34].
Phage displayed mimotopes can be used to characterize antigen-specific polysera
In 1994, Folgori et al. employed a library of phage-displayed random peptides to characterize antibody specificities in polyserum from patients vaccinated with Hepatitis B virus surface antigen (HBsAg) [35]. They identified mimotopes of two different epitopes within HBsAg. Sera from 20 different vaccinees displayed reactivity for these mimotopes, while sera of non-immune individuals failed to bind. This study opened a new approach to diagnostics and vaccine development based on phage display epitope library screening. Similarly, random peptide libraries were screened with polysera from patients with Lyme disease and 17 peptides were selected that distinguished patients with Lyme disease from healthy controls, demonstrating the value of this technology for developing serological diagnostic tests [36].
The phage display technology has been used to investigate the nature of anti-HIV antibodies present in polyserum from long-term non-progressors (LTNPs). These individuals typically possess antibodies with broad neutralization [37, 38, 39]. Understanding the epitope targets of these broadly neutralizing antibodies could provide important information for developing diagnostic tools and vaccination strategies. In the first study of this kind, Scala et al. identified ten mimotopes, which had wide cross-reactivity with polysera from LTNPs and SHIV-infected monkeys [40]. The phage-borne epitopes were immunogenic and four out of five monkeys immunized with these phages experienced lower levels of peak viremia following infection [40], supporting the use of this technology for vaccine development. Another study was performed with polyserum from a single LTNP patient, which identified a single epitope located in gp41 protein. Probing the isolated phages with variety of sera revealed that different sera have different affinity to mimotope variants (some sera did not bind some of the variants at all) thus demonstrating the diversity of the immune response [32]. This diversity likely reflects variability among the circulating virus strains within the regions that are targeted by antibodies; understanding the relationship between this diversity and effective neutralization will likely provide novel insight into the mechanisms of neutralization.
Since mimotopes generally display only small homologies with the target protein, large numbers of unique mimotopes are required to properly analyze complex polysera. The Dietrich group reported a study wherein ~700 mimotopes were selected by screening a phage display epitope library with eight LTNP patient sera [41]. Some of the mimotope sequences were attributed to HIV antigens based on linear homology and conformational similarities. However, it remains unclear whether the predicted epitopes represent targets for neutralization since immunization of mice with mimotopes selected from their phage collection only produced modest HIV-specific antibody responses with low neutralizing potential. Therefore, although this technology offers great promise for serological diagnostics, as in the case of Hepatitis B virus vaccinees and patients afflicted with Lyme disease, further refinement is required to employ this methodology for discerning specific neutralizing epitopes, as in the case of LTNPs.
Algorithms for deciphering epitopes using collections of affinity-selected mimotopes
In terms of characterizing polyserum using this methodology, our ultimate goal is to employ the mimotopes to define specific epitopes. While this has been done successfully for monoclonal antibodies [11, 32, 42, 43] where the target antigen is well-defined, this process is substantially more complicated in the case of polyclonal sera where a constellation of antigens are targeted. A common strategy for epitope identification involves simple homology searches where the linear sequence of the peptide is aligned with the corresponding linear sequence of a putative antigen [41]. However, this approach is applicable only for linear, or partially-conformational epitopes, which consist of small linear fragments. Strategies based on linear homology cannot be applied to genuine conformational epitopes since very few residues will be proximal to each other in the linear sequence. Additionally, we have observed that application of the linear alignment method identifies short linear fragments (typically 3-5 amino acids) that appear to be specific but are actually non-specific as they can also be found in the irrelevant proteins that we use for negative controls (Denisov, Denisova and Bramson, unpublished data). As an example, the sequence TPPG was uncovered as a common linear element within phage-displayed mimotopes isolated with polyserum from West Nile-infected individuals and this sequence maps equally well to great numbers of non-viral protein sequences taken from the NCBI protein database. Similar results were obtained with other short homologies like PAS, RSLT and RRP. The mimotopes containing these fragments can be attributed to many proteins suggesting that they likely represent common backbone motifs that provide a basic structure to the peptide for low-affinity antibody binding and they complicate unambiguous epitope mapping using linear homologies. Similar observations have been made by other groups [44] where turn-like structures for the sequences DVQX, XPGS, DITX, and DXSF were conserved between specific epitopes and a number of unrelated proteins, suggesting that these linear epitopes have inherent conformational preferences.
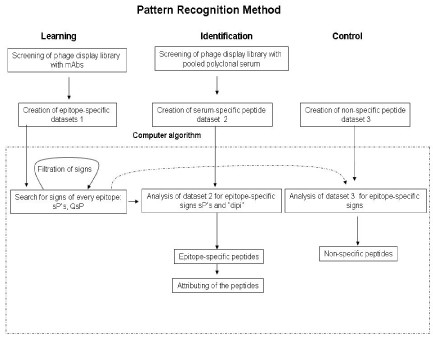
Scheme of computer algorithm based on pattern recognition theory. Signs – amino acid pairs chosen from different positions within an epitope sequence . Dipi – discrimination parameter equal to total number of epitope specific “signs” found at learning step. sP – “space pairs” - amino acid pairs separated in a peptide by one, two, three amino acids . QsP - is the quality of sP which is defined by the occurrence of the particular sP in all epitope mimotopes at learning.
Employing mimotopes as the basis for epitope-specific vaccination
While the mimotope strategy offers a powerful tool for monitoring epitope-specific antibodies following vaccination or infection, these mimetics may also provide a useful basis for the development of epitope-specific vaccines. A critical parameter in this regard is the degree to which the mimotope accurately mimics the cognate epitope. Some peptides selected from phage-display libraries by antibodies were used to successfully elicit cross-reactive antibody responses in animal models [53, 54]; in some cases, these antibody responses have conferred neutralization in vitro [55] and protection [56]. However, many mimotope peptides failed to induce antibodies cross-reactive with corresponding antigens [57, 58, 59, 60]. The issues related to the inability of mimotopes to elicit antibodies that were reactive to the native antigen were elegantly explored in the case of the B2.1 mimotope which was isolated using the anti-HIV neutralizing antibody, b12 [61, 62]. Although B2.1 displayed high affinity for b12, immunization of mice and rabbits with B2.1 failed to elicit HIV-specific antibodies. The crystal structure of b12 complexed with B2.1 was solved and compared to the structure of b12 bound to gp120 of HIV [61]. This comparison revealed that, although the binding in both cases was specific, the mimotope contacted different residues on b12 than gp120. These data reveal that although affinity selected peptides are antibody-specific, they are not always true mimetics of the antigenic epitope. While we, and others, commonly refer to immunoaffinity-selected peptides as mimotopes on the basis of their specific ability to bind antibody, it may be more appropriate to define them as binding site-specific peptides. It is clear that some of these peptides can display high-affinity interactions with the antibody binding site in way that is distinct from the true epitope on the target protein. The antibody binding site, which is formed by six hypervariable CDR loops, forms a continuous surface approximately 2800 A2 in area that is responsible for antigen binding according to X-ray crystallographic analysis. Yet, only a fraction of the CDR surface is found to constitute the combining site. As an example, for the monoclonal antibodies D1.3, HyHEL-5, HyHEL-lO, NC4I, and Je142, the surface that is used in the binding to antigen represents only 21%, 27%, 28%, 32%, and 22%, respectively, of the total surface formed by the CDRs. Similarly, the CDR residues that contact the antigen represent only 25%, 37%, 36%, 33%, and 27%, respectively, of the total number of CDR residues. It is not surprising, then, that when screening a complex library of random peptides, some peptides will be identified which bind to regions within the binding site that are not used to bind the target antigen. In this respect, monoclonal antibodies can actually be considered polyspecific [63]. Indeed, screening of peptide libraries with a monoclonal antibody typically yields a collection of different peptides that all have high affinity binding [31, 32, 64, 65, 66]. Yet, upon simple examination, these peptides do not have significant similarity to each other despite the fact that they should share some of appropriately located contact residues to be able to bind the antibody. Additionally, in view of the difference between sizes of epitope (average ~20 amino acids) and phage inserts, the mimotopes could mimic some aspects of epitope and different mimotopes could have partially overlapping sequences. The second reason for inability of mimotopes to induce cross-reactive immune response could be difference in a shape of binding site of antibodies induced by native antigen and mimotopes. These shapes are determined by the shape of the antigen and it could be different for a flat protein surface and extended peptides [67]. During affinity maturation antibody binding site acquires the best complementarities for the peptide antigen and thus evolves from the best fit for protein antigen binding. We believe that developing effective mimotope vaccines will require detailed structural analysis of the antibody binding to the target antigen to ensure that the best mimetics are selected in order to use them for the development of epitope-specific vaccines.
Conclusion
Screening of a phage display library with polyclonal serum is technically simple and amenable to routine laboratory analysis. This technology permits fine characterization of serum detecting not only antibodies directed to different domains of a protein but also antibodies directed to specific epitopes [68]. Using this methodology, it is possible to develop a characteristic “antibody signature” that can distinguish between different polysera directed at the same antigen [69]. Our results have demonstrated that the pattern recognition algorithm can effectively be employed to screen a mixture of antibodies and define the breadth of epitopes recognized by polyserum directed against specific proteins. The method can be further improved with a larger collection of mimotopes to be used in the learning step of the algorithm. “Next Generation Sequencing” technology, which allows sequencing of one gigabase of DNA in a couple of days [70, 71], might be beneficial in this regard, as it can enable, presumably, the sequencing of millions of phage in a single run. In principle, our pattern recognition method could also be applied to cases when there are no available monoclonal antibodies by using computational strategies to predict antibody epitopes. Multiple algorithms have been designed for prediction of antibody epitopes using atomic coordinates of the antigen or simple amino acid sequences [48][72, 73, 74]. For example, it is known that binding sites on a surface of proteins (commonly referred to as “hot spots”) have preferential amino acid composition, secondary structures and packing density. Amino acids as tryptophan, arginine and tyrosine were shown to be enriched in these hot spots [75]. We used one of these approaches [73] for analysis of polyclonal immune response to HER-2 protein and found that our predictive algorithm gave the outcome similar to theoretically predicted epitopes [69]. Based on this information and the algorithm described in the current review, we are developing novel software tools that can predict all hypothetical protein antigenic sites and present as sets of amino acid pairs, which can subsequently be employed as epitope signs for use with our mimotope recognition computer algorithm.
While the selected mimotopes may prove useful in the development of epitope-specific vaccines, the current state of the art still relies on a high degree of chance in the selection of the mimotope. To facilitate mimotope selection for vaccine applications, highly diverse libraries should be used for screening of polyclonal neutralizing serum and large collections of mimotopes should be selected. Computer assisted analysis of these large collections can help to select mimotopes enriched with epitope specific signs and scored according to the discrimination parameter. Should this selection method fail to reproducibly identify useful mimetics, it may be necessary to combine our bioinformatics strategies with structural analysis to identify “best fit” peptides.
List of abbreviations
- HBsAg:
-
Hepatitis B virus surface antigen
- LTNP:
-
long-term non-progressor
- E protein:
-
envelope protein
- CDR:
-
complementarity determining regions
- Signs:
-
amino acid pairs chosen from different positions within an epitope sequence
- Dipi:
-
discrimination parameter equal to total number of epitope specific “signs” found at learning step
- sP:
-
“space pairs” - amino acid pairs separated in a peptide by one, two, three amino acids
- QsP:
-
the quality of sP which is defined by the occurrence of the particular sP in all epitope mimotopes at learning.
Declarations
Acknowledgements
We are grateful for research support from the Canadian Breast Cancer Research Alliance, the National Institutes of Health USA, and the Ontario Institute for Cancer Research.
This article has been published as part of Immunome Research Volume 6 Supplement 2, 2010: Computational Vaccinology: State-of-the-art Assessments. The full contents of the supplement are available online at http://www.immunome-research.com/supplements/6/S2.
Authors’ Affiliations
References
- Amanna IJ, Carlson NE, Slifka MK: Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007, 357: 1903-15. 10.1056/NEJMoa066092.View ArticlePubMedGoogle Scholar
- Oliphant T, Nybakken GE, Engle M, Xu Q, Nelson CA, Sukupolvi-Petty S, Marri A, Lachmi BE, Olshevsky U, Fremont DH, et al: Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J Virol. 2006, 80: 12149-59. 10.1128/JVI.01732-06.PubMed CentralView ArticlePubMedGoogle Scholar
- Yip YL, Smith G, Koch J, Dubel S, Ward RL: Identification of epitope regions recognized by tumor inhibitory and stimulatory anti-ErbB-2 monoclonal antibodies: implications for vaccine design. J Immunol. 2001, 166: 5271-8.View ArticlePubMedGoogle Scholar
- Stancovski I, Hurwitz E, Leitner O, Ullrich A, Yarden Y, Sela M: Mechanistic aspects of the opposing effects of monoclonal antibodies to the ERBB2 receptor on tumor growth. Proc Natl Acad Sci U S A. 1991, 88: 8691-5. 10.1073/pnas.88.19.8691.PubMed CentralView ArticlePubMedGoogle Scholar
- Kiyamova R, Kostianets O, Malyuchik S, Filonenko V, Usenko V, Gurtovyy V, Khozayenko Y, Antonuk S, Old L, Gout I: Identification of Tumor-Associated Antigens from Medullary Breast Carcinoma by a Modified SEREX Approach. Mol Biotechnol. 2010Google Scholar
- Boutell JM, Hart DJ, Godber BL, Kozlowski RZ, Blackburn JM: Functional protein microarrays for parallel characterisation of p53 mutants. Proteomics. 2004, 4: 1950-8. 10.1002/pmic.200300722.View ArticlePubMedGoogle Scholar
- Blackburn JM, Hart DJ: Fabrication of protein function microarrays for systems-oriented proteomic analysis. Methods Mol Biol. 2005, 310: 197-216. full_text.View ArticlePubMedGoogle Scholar
- Chen YT: Cancer vaccine: identification of human tumor antigens by SEREX. Cancer J. 2000, 6 (Suppl 3): S208-17.PubMedGoogle Scholar
- Obata Y, Takahashi T, Sakamoto J, Tamaki H, Tominaga S, Hamajima N, Chen YT, Old LJ: SEREX analysis of gastric cancer antigens. Cancer Chemother Pharmacol. 2000, 46 (Suppl): S37-42. 10.1007/PL00014048.View ArticlePubMedGoogle Scholar
- Zvirbliene A, Kucinskaite I, Sezaite I, Samuel D, Sasnauskas K: Mapping of B cell epitopes in measles virus nucleocapsid protein. Arch Virol. 2007, 152: 25-39. 10.1007/s00705-006-0837-5.View ArticlePubMedGoogle Scholar
- Fernandez-Alonso M, Lorenzo G, Perez L, Bullido R, Estepa A, Lorenzen N, Coll JM: Mapping of linear antibody epitopes of the glycoprotein of VHSV, a salmonid rhabdovirus. Dis Aquat Organ. 1998, 34: 167-76. 10.3354/dao034167.View ArticlePubMedGoogle Scholar
- Van Regenmortel MHV: Mapping Epitope Structure and Activity: From One-Dimensional Prediction to Four-Dimensional Description of Antigenic Specificity. Methods. 1996, 9: 465-72. 10.1006/meth.1996.0054.View ArticlePubMedGoogle Scholar
- Geysen HM, Rodda SJ, Mason TJ: A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol Immunol. 1986, 23: 709-15. 10.1016/0161-5890(86)90081-7.View ArticlePubMedGoogle Scholar
- Kopple KD, Go A, Pilipauskas DR: Studies of peptide conformation. Evidence for beta structures in solutions of linear tetrapeptides containing proline. J Am Chem Soc. 1975, 97: 6830-8. 10.1021/ja00856a039.View ArticlePubMedGoogle Scholar
- Sidhu SS: Engineering M13 for phage display. Biomol Eng. 2001, 18: 57-63. 10.1016/S1389-0344(01)00087-9.View ArticlePubMedGoogle Scholar
- Kenrick SA, Daugherty PS: Bacterial display enables efficient and quantitative peptide affinity maturation. Protein Eng Des Sel. 2010, 23: 9-17. 10.1093/protein/gzp065.PubMed CentralView ArticlePubMedGoogle Scholar
- Pershad K, Pavlovic JD, Graslund S, Nilsson P, Colwill K, Karatt-Vellatt A, Schofield DJ, Dyson MR, Pawson T, Kay BK, et al: Generating a panel of highly specific antibodies to 20 human SH2 domains by phage display. Protein Eng Des Sel. 2010, 23: 279-88. 10.1093/protein/gzq003.PubMed CentralView ArticlePubMedGoogle Scholar
- Shibasaki S, Maeda H, Ueda M: Molecular display technology using yeast--arming technology. Anal Sci. 2009, 25: 41-9. 10.2116/analsci.25.41.View ArticlePubMedGoogle Scholar
- Kondo A, Ueda M: Yeast cell-surface display--applications of molecular display. Appl Microbiol Biotechnol. 2004, 64: 28-40. 10.1007/s00253-003-1492-3.View ArticlePubMedGoogle Scholar
- Yan X, Xu Z: Ribosome-display technology: applications for directed evolution of functional proteins. Drug Discov Today. 2006, 11: 911-6. 10.1016/j.drudis.2006.08.012.View ArticlePubMedGoogle Scholar
- Rothe A, Hosse RJ, Power BE: Ribosome display for improved biotherapeutic molecules. Expert Opin Biol Ther. 2006, 6: 177-87. 10.1517/14712598.6.2.177.View ArticlePubMedGoogle Scholar
- He M, Khan F: Ribosome display: next-generation display technologies for production of antibodies in vitro. Expert Rev Proteomics. 2005, 2: 421-30. 10.1586/14789450.2.3.421.View ArticlePubMedGoogle Scholar
- Matsumura N, Tsuji T, Sumida T, Kokubo M, Onimaru M, Doi N, Takashima H, Miyamoto-Sato E, Yanagawa H: mRNA display selection of a high-affinity, Bcl-X(L)-specific binding peptide. FASEB J. 2010, 24: 2201-10. 10.1096/fj.09-143008.View ArticlePubMedGoogle Scholar
- Roberts RW, Szostak JW: RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997, 94: 12297-302. 10.1073/pnas.94.23.12297.PubMed CentralView ArticlePubMedGoogle Scholar
- Zapun A, Bardwell JC, Creighton TE: The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993, 32: 5083-92. 10.1021/bi00070a016.View ArticlePubMedGoogle Scholar
- Luzzago A, Felici F, Tramontano A, Pessi A, Cortese R: Mimicking of discontinuous epitopes by phage-displayed peptides, I. Epitope mapping of human H ferritin using a phage library of constrained peptides. Gene. 1993, 128: 51-7. 10.1016/0378-1119(93)90152-S.View ArticlePubMedGoogle Scholar
- Felici F, Luzzago A, Folgori A, Cortese R: Mimicking of discontinuous epitopes by phage-displayed peptides, II. Selection of clones recognized by a protective monoclonal antibody against the Bordetella pertussis toxin from phage peptide libraries. Gene. 1993, 128: 21-7. 10.1016/0378-1119(93)90148-V.View ArticlePubMedGoogle Scholar
- Pirofski LA: Polysaccharides, mimotopes and vaccines for fungal and encapsulated pathogens. Trends Microbiol. 2001, 9: 445-51. 10.1016/S0966-842X(01)02134-5.View ArticlePubMedGoogle Scholar
- Beenhouwer DO, May RJ, Valadon P, Scharff MD: High affinity mimotope of the polysaccharide capsule of Cryptococcus neoformans identified from an evolutionary phage peptide library. J Immunol. 2002, 169: 6992-9.View ArticlePubMedGoogle Scholar
- Sidhu SS, Feld BK, Weiss GA: M13 bacteriophage coat proteins engineered for improved phage display. Methods Mol Biol. 2007, 352: 205-19.PubMedGoogle Scholar
- Denisova GF, Denisov DA, Yeung J, Loeb MB, Diamond MS, Bramson JL: A novel computer algorithm improves antibody epitope prediction using affinity-selected mimotopes: a case study using monoclonal antibodies against the West Nile virus E protein. Mol Immunol. 2008, 46: 125-34. 10.1016/j.molimm.2008.07.020.View ArticlePubMedGoogle Scholar
- Enshell-Seijffers D, Denisov D, Groisman B, Smelyanski L, Meyuhas R, Gross G, Denisova G, Gershoni JM: The mapping and reconstitution of a conformational discontinuous B-cell epitope of HIV-1. J Mol Biol. 2003, 334: 87-101. 10.1016/j.jmb.2003.09.002.View ArticlePubMedGoogle Scholar
- Odorico M, Pellequer JL: BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J Mol Recognit. 2003, 16: 20-2. 10.1002/jmr.602.View ArticlePubMedGoogle Scholar
- Blythe MJ, Flower DR: Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 2005, 14: 246-8. 10.1110/ps.041059505.PubMed CentralView ArticlePubMedGoogle Scholar
- Folgori A, Tafi R, Meola A, Felici F, Galfre G, Cortese R, Monaci P, Nicosia A: A general strategy to identify mimotopes of pathological antigens using only random peptide libraries and human sera. EMBO J. 1994, 13: 2236-43.PubMed CentralPubMedGoogle Scholar
- Kouzmitcheva GA, Petrenko VA, Smith GP: Identifying diagnostic peptides for lyme disease through epitope discovery. Clin Diagn Lab Immunol. 2001, 8: 150-60.PubMed CentralPubMedGoogle Scholar
- Zhang YJ, Fracasso C, Fiore JR, Bjorndal A, Angarano G, Gringeri A, Fenyo EM: Augmented serum neutralizing activity against primary human immunodeficiency virus type 1 (HIV-1) isolates in two groups of HIV-1-infected long-term nonprogressors. J Infect Dis. 1997, 176: 1180-7. 10.1086/514111.View ArticlePubMedGoogle Scholar
- Pilgrim AK, Pantaleo G, Cohen OJ, Fink LM, Zhou JY, Zhou JT, Bolognesi DP, Fauci AS, Montefiori DC: Neutralizing antibody responses to human immunodeficiency virus type 1 in primary infection and long-term-nonprogressive infection. J Infect Dis. 1997, 176: 924-32. 10.1086/516508.View ArticlePubMedGoogle Scholar
- Wang Q, Shang H, Han X, Zhang Z, Jiang Y, Wang Y, Dai D, Diao Y: High level serum neutralizing antibody against HIV-1 in Chinese long-term non-progressors. Microbiol Immunol. 2008, 52: 209-15. 10.1111/j.1348-0421.2008.00033.x.View ArticlePubMedGoogle Scholar
- Chen X, Scala G, Quinto I, Liu W, Chun TW, Justement JS, Cohen OJ, vanCott TC, Iwanicki M, Lewis MG, et al: Protection of rhesus macaques against disease progression from pathogenic SHIV-89.6PD by vaccination with phage-displayed HIV-1 epitopes. Nat Med. 2001, 7: 1225-31. 10.1038/nm1101-1225.View ArticlePubMedGoogle Scholar
- Humbert M, Antoni S, Brill B, Landersz M, Rodes B, Soriano V, Wintergerst U, Knechten H, Staszewski S, von Laer D, et al: Mimotopes selected with antibodies from HIV-1-neutralizing long-term non-progressor plasma. Eur J Immunol. 2007, 37: 501-15. 10.1002/eji.200636560.View ArticlePubMedGoogle Scholar
- Paley EL, Smelyanski L, Malinovskii V, Subbarayan PR, Berdichevsky Y, Posternak N, Gershoni JM, Sokolova O, Denisova G: Mapping and molecular characterization of novel monoclonal antibodies to conformational epitopes on NH2 and COOH termini of mammalian tryptophanyl-tRNA synthetase reveal link of the epitopes to aggregation and Alzheimer's disease. Mol Immunol. 2007, 44: 541-57. 10.1016/j.molimm.2006.02.006.View ArticlePubMedGoogle Scholar
- Hu S, Zhu Z, Li L, Chang L, Li W, Cheng L, Teng M, Liu J: Epitope mapping and structural analysis of an anti-ErbB2 antibody A21: Molecular basis for tumor inhibitory mechanism. Proteins. 2008, 70: 938-49. 10.1002/prot.21551.View ArticlePubMedGoogle Scholar
- Craig L, Sanschagrin PC, Rozek A, Lackie S, Kuhn LA, Scott JK: The role of structure in antibody cross-reactivity between peptides and folded proteins. J Mol Biol. 1998, 281: 183-201. 10.1006/jmbi.1998.1907.View ArticlePubMedGoogle Scholar
- Denisov DA, Denisova GF, Lelic A, Loeb MB, Bramson JL: Deciphering epitope specificities within polyserum using affinity selection of random peptides and a novel algorithm based on pattern recognition theory. Mol Immunol. 2009, 46: 429-36. 10.1016/j.molimm.2008.10.013.View ArticlePubMedGoogle Scholar
- Denisov DA, Drozdov-Tichomirov LN, Grigoryeva DN: Pattern recognition methods for prediction of helical region in proteins. J Theor Biol. 1973, 41: 431-9. 10.1016/0022-5193(73)90053-2.View ArticlePubMedGoogle Scholar
- Bongard M: Pattern Recognition. 1970, Spartan Books, Rochelle Park, NJGoogle Scholar
- Greenbaum JA, Andersen PH, Blythe M, Bui HH, Cachau RE, Crowe J, Davies M, Kolaskar AS, Lund O, Morrison S, et al: Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J Mol Recognit. 2007, 20: 75-82. 10.1002/jmr.815.View ArticlePubMedGoogle Scholar
- Menendez A, Scott JK: The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal Biochem. 2005, 336: 145-57. 10.1016/j.ab.2004.09.048.View ArticlePubMedGoogle Scholar
- Tchernychev B, Cabilly S, Wilchek M: The epitopes for natural polyreactive antibodies are rich in proline. Proc Natl Acad Sci U S A. 1997, 94: 6335-9. 10.1073/pnas.94.12.6335.PubMed CentralView ArticlePubMedGoogle Scholar
- Kay BK, Williamson MP, Sudol M: The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14: 231-41.PubMedGoogle Scholar
- Ravi Chandra B, Gowthaman R, Raj Akhouri R, Gupta D, Sharma A: Distribution of proline-rich (PxxP) motifs in distinct proteomes: functional and therapeutic implications for malaria and tuberculosis. Protein Eng Des Sel. 2004, 17: 175-82. 10.1093/protein/gzh024.View ArticlePubMedGoogle Scholar
- Puntoriero G, Meola A, Lahm A, Zucchelli S, Ercole BB, Tafi R, Pezzanera M, Mondelli MU, Cortese R, Tramontano A, et al: Towards a solution for hepatitis C virus hypervariability: mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO J. 1998, 17: 3521-33. 10.1093/emboj/17.13.3521.PubMed CentralView ArticlePubMedGoogle Scholar
- Mattioli S, Imberti L, Stellini R, Primi D: Mimicry of the immunodominant conformation-dependent antigenic site of hepatitis A virus by motifs selected from synthetic peptide libraries. J Virol. 1995, 69: 5294-9.PubMed CentralPubMedGoogle Scholar
- Chargelegue D, Obeid OE, Hsu SC, Shaw MD, Denbury AN, Taylor G, Steward MW: A peptide mimic of a protective epitope of respiratory syncytial virus selected from a combinatorial library induces virus-neutralizing antibodies and reduces viral load in vivo. J Virol. 1998, 72: 2040-6.PubMed CentralPubMedGoogle Scholar
- Scala G, Chen X, Liu W, Telles JN, Cohen OJ, Vaccarezza M, Igarashi T, Fauci AS: Selection of HIV-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. J Immunol. 1999, 162: 6155-61.PubMedGoogle Scholar
- Ho J, Uger RA, Zwick MB, Luscher MA, Barber BH, MacDonald KS: Conformational constraints imposed on a pan-neutralizing HIV-1 antibody epitope result in increased antigenicity but not neutralizing response. Vaccine. 2005, 23: 1559-73. 10.1016/j.vaccine.2004.09.037.View ArticlePubMedGoogle Scholar
- El Kasmi KC, Fillon S, Theisen DM, Hartter H, Brons NH, Muller CP: Neutralization of measles virus wild-type isolates after immunization with a synthetic peptide vaccine which is not recognized by neutralizing passive antibodies. J Gen Virol. 2000, 81 (*): 729-35.View ArticlePubMedGoogle Scholar
- El Kasmi KC, Deroo S, Theisen DM, Brons NH, Muller CP: Crossreactivity of mimotopes and peptide homologues of a sequential epitope with a monoclonal antibody does not predict crossreactive immunogenicity. Vaccine. 1999, 18: 284-90. 10.1016/S0264-410X(99)00199-1.View ArticlePubMedGoogle Scholar
- Van Regenmortel MH: Reductionism and complexity in molecular biology. Scientists now have the tools to unravel biological and overcome the limitations of reductionism. EMBO Rep. 2004, 5: 1016-20. 10.1038/sj.embor.7400284.View ArticlePubMedGoogle Scholar
- Saphire EO, Montero M, Menendez A, van Houten NE, Irving MB, Pantophlet R, Zwick MB, Parren PW, Burton DR, Scott JK, et al: Structure of a high-affinity "mimotope" peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J Mol Biol. 2007, 369: 696-709. 10.1016/j.jmb.2007.01.060.PubMed CentralView ArticlePubMedGoogle Scholar
- Zwick MB, Bonnycastle LL, Menendez A, Irving MB, Barbas CF, Parren PW, Burton DR, Scott JK: Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J Virol. 2001, 75: 6692-9. 10.1128/JVI.75.14.6692-6699.2001.PubMed CentralView ArticlePubMedGoogle Scholar
- Van Regenmortel MH: From absolute to exquisite specificity. Reflections on the fuzzy nature of species, specificity and antigenic sites. J Immunol Methods. 1998, 216: 37-48. 10.1016/S0022-1759(98)00069-6.View ArticlePubMedGoogle Scholar
- Denisova G, Stern B, Raviv D, Zwickel J, Smorodinsky NI, Gershoni JM: Humoral immune response to immunocomplexed HIV envelope glycoprotein 120. AIDS Res Hum Retroviruses. 1996, 12: 901-9. 10.1089/aid.1996.12.901.View ArticlePubMedGoogle Scholar
- Boots LJ, McKenna PM, Arnold BA, Keller PM, Gorny MK, Zolla-Pazner S, Robinson JE, Conley AJ: Anti-human immunodeficiency virus type 1 human monoclonal antibodies that bind discontinuous epitopes in the viral glycoproteins can identify mimotopes from recombinant phage peptide display libraries. AIDS Res Hum Retroviruses. 1997, 13: 1549-59. 10.1089/aid.1997.13.1549.View ArticlePubMedGoogle Scholar
- Bublil EM, Yeger-Azuz S, Gershoni JM: Computational prediction of the cross-reactive neutralizing epitope corresponding to the [corrected] monclonal [corrected] antibody b12 specific for HIV-1 gp120. FASEB J. 2006, 20: 1762-74. 10.1096/fj.05-5509rev.View ArticlePubMedGoogle Scholar
- MacCallum RM, Martin AC, Thornton JM: Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol. 1996, 262: 732-45. 10.1006/jmbi.1996.0548.View ArticlePubMedGoogle Scholar
- Folgori A, Luzzago A, Monaci P, Nicosia A, Cortese R, Felici F: Identification of disease-specific epitopes. Methods Mol Biol. 1998, 87: 195-208.PubMedGoogle Scholar
- Denisova G, Denisov D, Evelegh C, Weissgram M, Beck J, Foley SR, Bramson JL: Characterizing complex polysera produced by antigen-specific immunization through the use of affinity-selected mimotopes. PLoS One. 2009, 4: e5309-10.1371/journal.pone.0005309.PubMed CentralView ArticlePubMedGoogle Scholar
- van Vliet AH: Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol Lett. 2010, 302: 1-7. 10.1111/j.1574-6968.2009.01767.x.View ArticlePubMedGoogle Scholar
- Metzker ML: Sequencing technologies - the next generation. Nat Rev Genet. 2010, 11: 31-46. 10.1038/nrg2626.View ArticlePubMedGoogle Scholar
- Roggen EL: Recent developments with B-cell epitope identification for predictive studies. J Immunotoxicol. 2006, 3: 137-49. 10.1080/15476910600845690.View ArticlePubMedGoogle Scholar
- Batori V, Friis EP, Nielsen H, Roggen EL: An in silico method using an epitope motif database for predicting the location of antigenic determinants on proteins in a structural context. J Mol Recognit. 2006, 19: 21-9. 10.1002/jmr.752.View ArticlePubMedGoogle Scholar
- Moreira IS, Fernandes PA, Ramos MJ: Hot spots--a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007, 68: 803-12. 10.1002/prot.21396.View ArticlePubMedGoogle Scholar
- Bahadur RP, Zacharias M: The interface of protein-protein complexes: analysis of contacts and prediction of interactions. Cell Mol Life Sci. 2008, 65: 1059-72. 10.1007/s00018-007-7451-x.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.