
Stacking and energetic contribution of aromatic islands at the binding interface of antibody proteins
- Proceedings
- Open Access
Stacking and energetic contribution of aromatic islands at the binding interface of antibody proteins
https://doi.org/10.1186/1745-7580-6-S1-S1
© Cao et al; licensee BioMed Central Ltd. 2010
- Published: 27 September 2010
Abstract
Background
The enrichment and importance of some aromatic residues, such as Tyr and Trp, have been widely noticed at the binding interfaces of antibodies from many experimental and statistical results, some of which were even identified as “hot spots” contributing significantly greater to the binding affinity than other amino acids. However, how these aromatic residues influence the immune binding still deserves further investigation. A large-scale examination was done regarding the local spatial environment around the interfacial Tyr or Trp residues. Energetic contribution of these Tyr and Trp residues to the binding affinity was then studied regarding 82 representative antibody interfaces covering 509 immune complexes from the PDB database and IMGT/3Dstructure-DB.
Results
The connectivity analysis of interfacial residues showed that Tyr and Trp tended to cluster into the spatial Aromatic Islands (AI) rather than being distributed randomly at the antibody interfaces. Out of 82 antibody-antigen complexes, 72% (59) interfaces were found to contain AI with more than 3 aromatic residues. The statistical test against an empirical distribution indicated that the existence of AI was significant in about 60% representative antibody interfaces. Secondly, the loss of solvent accessible surface area (SASA) for side chains of aromatic residues between actually crowded state and independent state was nicely correlated with the AI size increasing in a linearly positive way which indicated that the aromatic side chains in AI tended to take a compact and ordered stacking conformation at the interfaces. Interestingly, the SASA loss of AI was also correlated roughly with the averaged gap of binding free energy between the theoretical and experimental data for immune complexes.
Conclusions
The results of our study revealed the wide existence and statistical significance of “Aromatic Island” (AI) composed of the spatially clustered Tyr and Trp residues at the antibody interfaces. The regular arrangement and stacking of aromatic side chains in AI could probably produce extra cooperative effects to the binding affinity which was firstly observed through the large-scale data analysis. The finding in this work not only provides insights into the functional role of aromatic residues in the antibody-antigen interaction, but also may facilitate the antibody engineering and potential clinical applications.
Keywords
- Aromatic Residue
- Solvent Accessible Surface Area
- Actual Interface
- Interfacial Residue
- Binding Interface
Background
It is well known that protein-protein interactions are fundamental to most of biological processes, including signal transduction, gene translation or transcription, enzyme activation or inhibition, and immune recognition. Contrast to the interaction between other normal protein-protein complexes, the binding between antibody and antigen is highly specific and stable [1]. Previous studies have revealed that this specificity is dominantly determined by the contacting interface which is mainly composed of the variable domains of antibody [2, 3, 4, 5, 6]. It has been reported that with only 5% sequence change in the variable domains, antibodies can recognize specifically and bind tightly to 1010 different antigens [7]. It is always interesting to study how antibody can recognize so large variety of antigens with so little change in sequence and thus deserve further investigation.
Characteristics of the binding interfaces of antibodies such as the size, shape, chemical, physical or structural complementation have been analyzed from different perspectives for a deeper understanding to antibody-antigen interactions [8, 9, 10]. Although the hydrophobic effect was considered as the major driving force for the general protein binding, the study of Tsai and co-workers indicated that hydrophobic amino acids were not the dominant part and a higher proportion of charged and polar residues could be found at the binding interfaces [11]. Subsequent comparison between the interfaces of six antibody-antigen complexes and other protein-protein complexes reported that the residues composing the interface of antibody-antigen complexes were more polar, protruding and accessible [12]. Currently, more and more results suggest that there are significant differences between the interfaces of immune and non-immune protein complexes. For instance, the interfaces of antigen-antibody complexes are particularly rich in Tyr, Arg, His, Phe and Trp [13, 14, 15, 16, 17]. Although further observations indicate that this enrichment ranking alters slightly with different data size, aromatic residues have always been found to occur more frequently at the binding sites of antibodies.
On the other hand, the contribution of enriched residues to the binding selectivity and specificity of antibody has aroused extensive interest [18]. By the virtue of alanine scanning mutagenesis, the energetic contribution of respective residue to protein binding could be evaluated with the observed free energy variation derived from the introduced mutation [19, 20, 21, 22]. The results of mutations have frequently indicated that the affinity change of mutating certain interfacial residue is far more unpredictable which is considered as the hot spot residue at the binding interface [23]. Some interfacial Tyr or Trp residues, but not all of them, have subsequently been identified as hot spots that contribute significantly to the high affinity of antibody-antigen interactions [24]. Despite that the different conclusions have been derived from several individual experiments adopting different datasets and methodologies, the enrichment and important role of Tyr and Trp residues have been widely noticed at the binding interfaces of antibodies.
However, several questions are still open to be answered. Why are these aromatic residues enriched and preferred? How do they affect the affinity so largely and form the “hot spots”? Are there any special local environment existing around the Tyr or Trp residues to facilitate the interaction at the interface? … In order to answer these questions, an in-depth and large-scale analysis would be helpful focusing on the aromatic residues at the binding interfaces of antibody-antigen complexes. Here, we conducted a comprehensive analysis of 82 non-redundant interfaces of antibodies covering 509 immune complexes from the PDB database [25] and IMGT/3Dstructure-DB [26, 27]. The residue connection and spatial distribution were scanned for all interfacial Tyr and Trp residues following an enrichment analysis. Systematic study was further focused on the relationship between the distribution pattern and the energetic contribution of aromatic interfacial residues in order to reveal the function of the aromatic residues in the binding interfaces of antibodies.
Results
“Aromatic Island” at the interface of antibodies
As to the antibody-antigen complexes collected in our dataset, the area of binding interfaces was firstly calculated. In general, the loss of solvent accessible surface area (SASA) was about 173~2351Å2 for antibodies upon the complex formation, and the average value was 644 Å2. The residue composition for interface of antibodies was also calculated. According to the absolute value of residue composition, Tyr, Ser, Thr were the top 3 most abundant residues at the interfacial area of antibodies with 17.10%, 14.61% and 8.66% respectively (Additional file 1). Removing the intrinsic abundance of residues in whole antibody, Tyr, Arg, and Trp were found to be the top 3 significantly enriched amino acids at the binding interface of antibodies (Additional file 1). Our finding is consistent with the previous results, especially for the significant enrichment of Tyr and Trp residues at the binding interface of antibodies [14, 15]. Different from Trp, Tyr residues are not only significantly enriched and preferred at the antibody interface, but also highly abundant for residue composition of antibody interface in the absolute value.
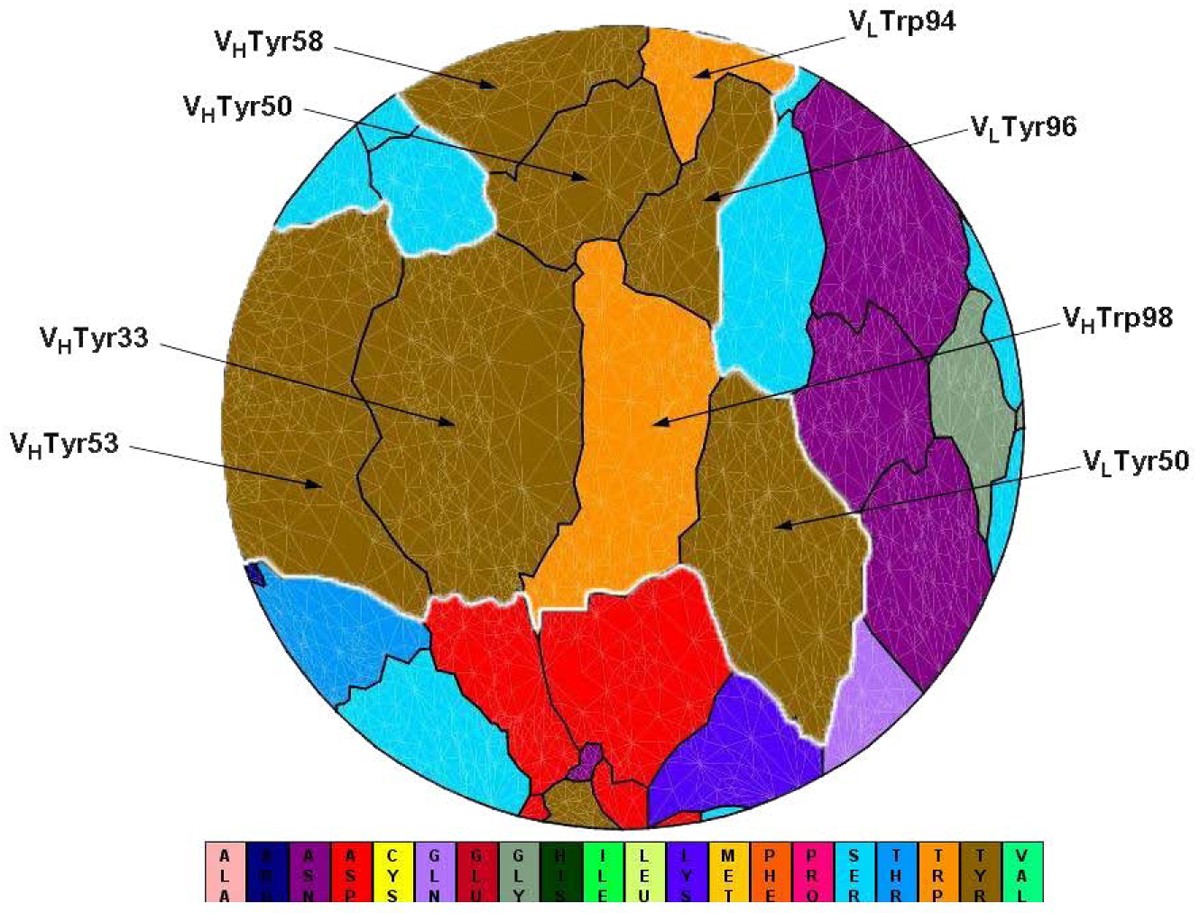
Flatten view of a binding interface at an antibody (PDB code 1c08). VLTyr50, VLTrp94, VLTyr96, VHTyr33, VHTyr50, VHTyr53, VHTyr58, VHTrp98 (IMGT numbering: V-Kappa Tyr56, V-Kappa Trp114, V-Kappa Tyr116, VH Tyr38, VH Tyr55, VH Tyr58, VH Tyr66, VH Trp107) cluster together to form a spatial “Aromatic Island” (abbreviated to “AI”) at the HyHEL-10 antibody interfaces, which is highlighted with white line (Tyr, colored in dark yellow; Trp, colored in orange). Figure 1 is generated by software JMiV.
Statistical analysis of “Aromatic Island”
Statistical Probability for Aromatic Island among simulative interfaces
Size of AIa |
Number of Antibody Interfaces |
Averaged Probabilityb |
Number of Interfaces with statistically significant AI |
Averaged Percentagec |
|---|---|---|---|---|
2 |
7 |
51.01% |
0 |
40.0% |
3 |
6 |
23.93% |
0 |
51.4% |
4 |
15 |
3.45% |
12 |
75.0% |
5 |
9 |
1.68% |
9 |
81.8% |
6 |
11 |
0.21% |
11 |
94.3% |
7 |
9 |
0.08% |
9 |
94.0% |
>= 8 |
8 |
0 |
8 |
94.6% |
Total |
65 |
/ |
49 |
/ |
Generally, the probability to observe AI among simulative interfaces dropped sharply from 51% to 0 with the AI size increasing from 2 to 12 residues. When the AI size is below 4 aromatic residues in actual interfaces, the probability for observing such AI was not statistically significant (the probability > 5.0%, statistical test against an empirical distribution). Furthermore, it was found that the statistical significance to observe AI was not only related to the AI size, but also related to the percentage of aromatic interfacial residues being clustered into AI. For example, there were 3 interfaces whose probabilities for observing AI were not statistically significant among 15 interfaces with AI size of 4 aromatic residues (Table 1). One reason could be that abundant aromatic residues were observed at the interface, but only less than 70% were included into AI.
Among all the 65 interfaces, 49 interfaces (75.4%) were found with significant AI when compared with the simulative interfaces (the probability < 5.0%, statistical test against an empirical distribution). In summary, the significant AI was observed at 60% of representative antibody interfaces (49/82). Thus, the majority of AI at the binding interfaces of antibodies were of statistical significance.
Regular arrangement of aromatic side chains in “Aromatic Island”
As we know that, both Tyr and Trp residues have bulky and rigid ring chain structure, comparing to most of other amino acids with either small or flexible side chains. According to previous research, the extending conformation would be preferred for an aromatic residue in open surrounding [28, 29]. How are the side chains arranged when so many aromatic rings crowd together in the AI at the binding interface of antibody? Since the compactness of side chains for aromatic residues in AI could be reflected by the loss between the sum of SASA for every side chain of aromatic residue in fully independent state and the actual SASA for AI in crowded state, we calculated the loss of SASA for every AI observed in all antibody interfaces. If the aromatic rings keep some compact and regular arrangement in AI, the loss of SASA would be correlated with AI size. Contrarily, the random arrangement of aromatic rings will not result in a nice correlation.
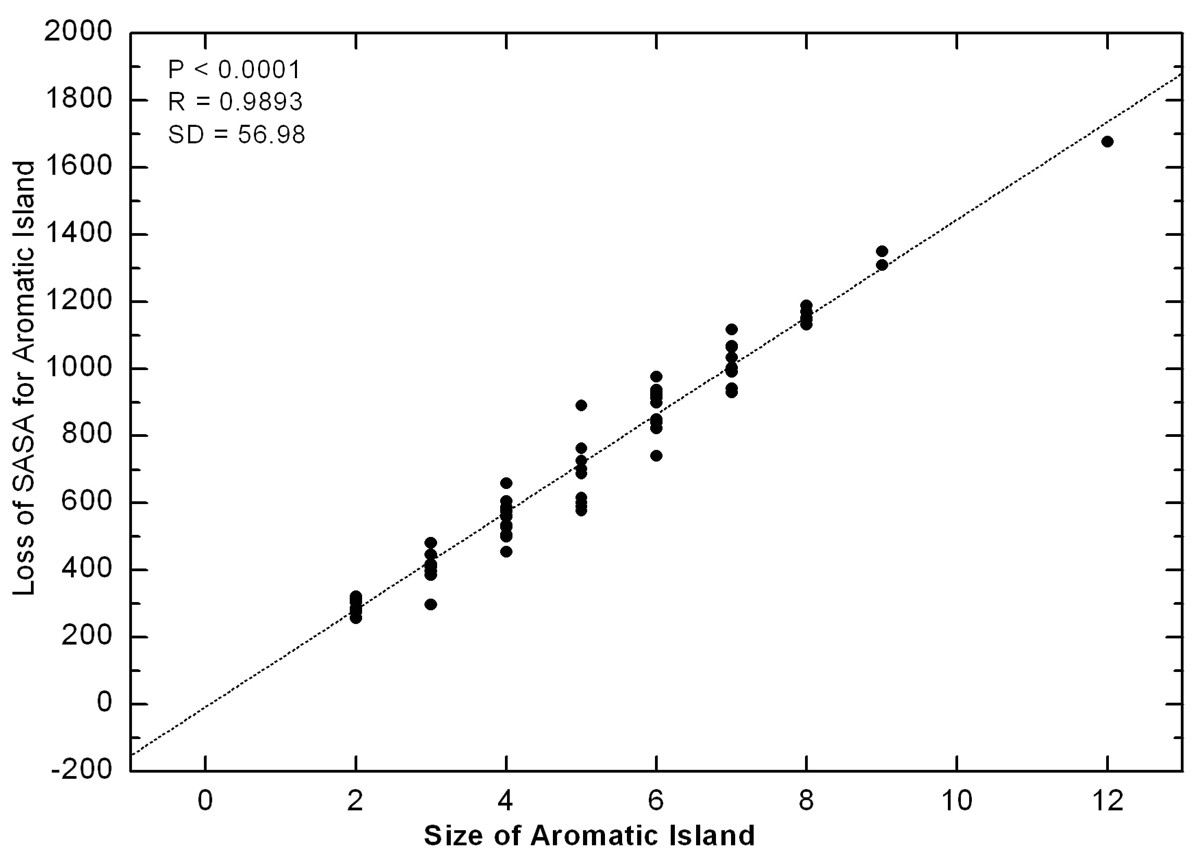
Correlation between the size of Aromatic Island and the loss of SASA for Aromatic Island The loss of SASA for AI (Å2) is calculated as the difference between the sum of SASA for every side chain of aromatic residue in fully independent state and the actual SASA for Aromatic Island. The size of Aromatic Island is recorded as the number of aromatic Tyr and Trp residues included in AI
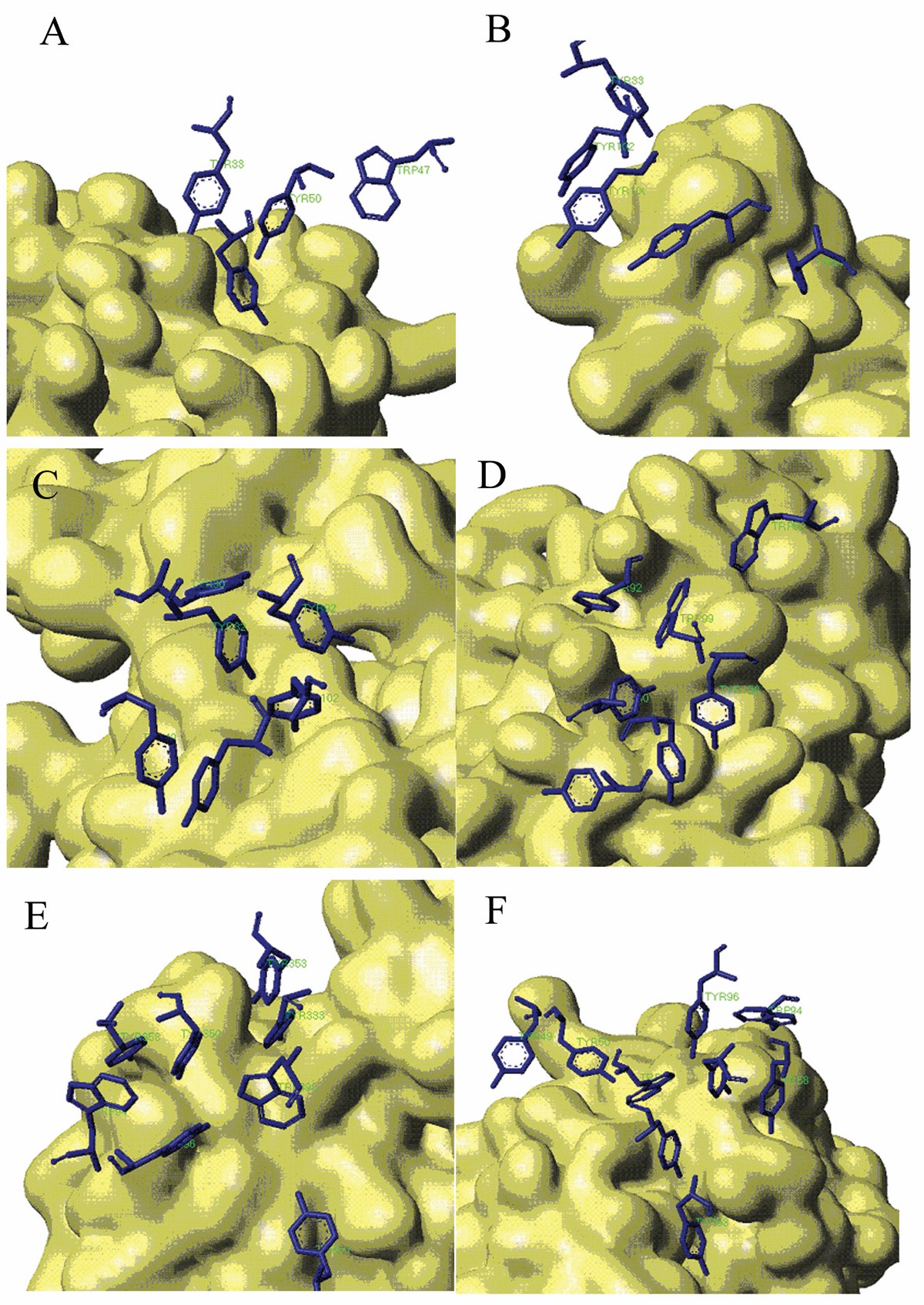
Stacking Conformation for aromatic residues in Aromatic Island The antigen protein is displayed with solvent surface mode. For better view, only the aromatic interfacial Tyr and Trp residues in AI are displayed with stick mode from the antibody. (A) Aromatic Island size of 4 at the binding interface of antibody 9D7 and IL-10 (PDB code 1lk3) (B) Aromatic Island size of 5 at the binding interface of antibody 33H1 and potassium channel molecule (PDB code 1ors) (C) Aromatic Island size of 6 at the binding interface of antibody and cytochrome AA3 (PDB code 1ar1) (D) Aromatic Island size of 7 at the binding interface of antibody YTS 105.18 and T-cell surface glycoprotein CD8 alpha chain (PDB code 2arj) (E) Aromatic Island size of 8 at the binding interface of antibody HyHEL-26 and HEL (PDB code 1ndm) (F) Aromatic Island size of 9 at the binding interface of antibody HyHEL-10 mutant and HEL (PDB code 2eiz)
Energetic contribution of “Aromatic Island” to binding affinity
It is well known that the abundant stacking of aromatic rings can produce strong and extra effects to stabilize the structures for nucleic acids [31, 32]. The regular stacking of aromatic side chains at antibody interface reminds us to verify whether such cooperative effect also exists during the binding of antibody and antigen.
Normally, it is realized that the experimental mutational data is the final effect observed. However, the computed data is actually a theoretical sum-up of individual mutation, where the cooperative effects between neighboring residues cannot be completely integrated. The experimental binding free energy for antibody-antigen complex is the result of global effect which covers the contribution of AI, while the computationally obtained binding free energy is usually the summary of individual residue effect. Under the same systemic error, the gap of binding free energy between the experimental and computational results might to some extent reflect the collective effect of AI if the stacking of aromatic rings does contribute to antibody-antigen binding.
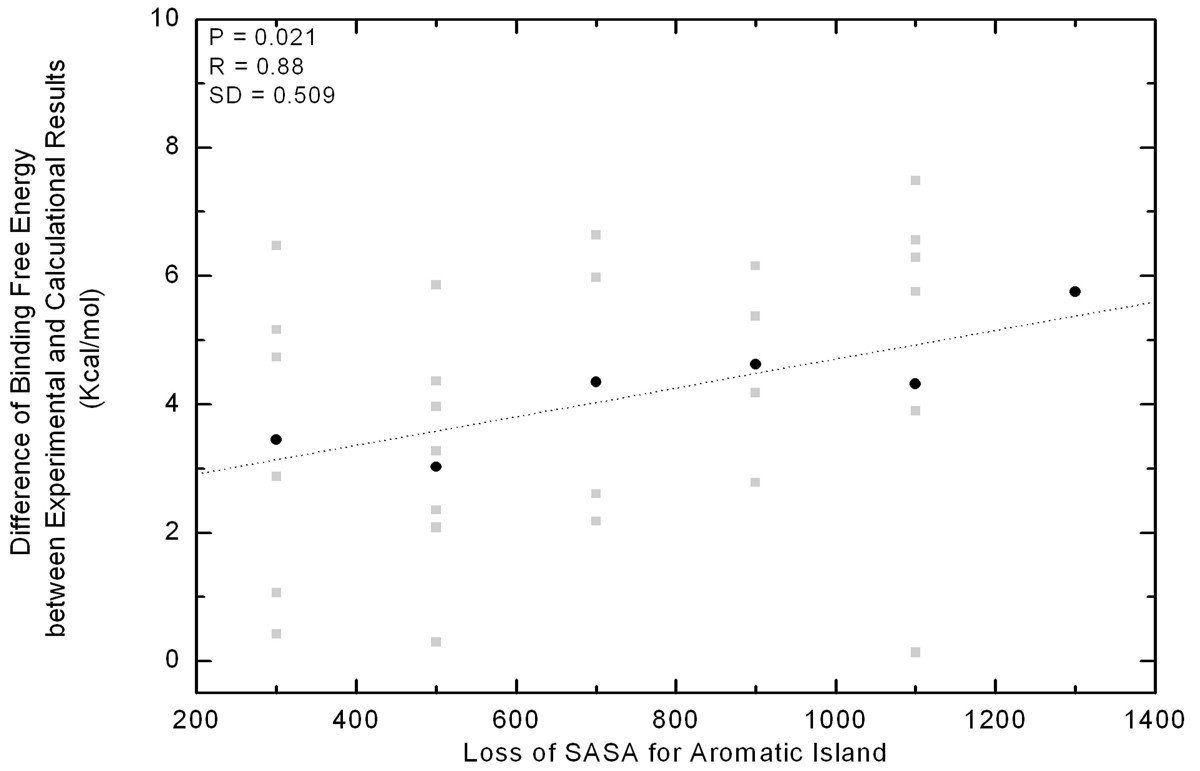
Correlation between the energetic gap and SASA loss of Aromatic Islands at 30 antibody interfaces. Along the x-axis all of the values in a bin (size 200 Å2) are pulled together as a group and shown in the middle. The gap of binding free energy between theoretical and experimental data is indicated with grey square for every immune complex. In each group, the gap is averaged and indicated with black dot.
Discussion
In this paper, the spatial distribution of enriched aromatic Tyr and Trp residues was investigated where compact and significant AI was widely found at most of the binding interfaces of antibodies. In previous experiments, several cases have been reported that Tyr or Trp plays an important role in various aspects, such as increasing the affinity and helping induce fit of antibody to antigen proteins [18, 19, 20, 21]. However, the utilization of individual residues seems not enough to fully explain the prevalence and enrichment of Tyr and Trp. The existence of AI at the antibody interfaces is acceptable to explain both above. Firstly, Tyr and Trp have been found to be able to form more close interactions to antigen proteins than other peers because of the aromatic and hydroxyl groups in their side chain [21, 28]. Interestingly, Phe is another amino acid with aromatic side chain, and was rarely found at the antibody interface. It is possibly because the absence of functional group in the aromatic ring.
Furthermore, the piling of aromatic rings may produce extra stabilizing energy for the whole structures of protein complexes, similar to the case in nucleic acids. In addition, the dense gathering of aromatic side chains could cause a highly hydrophobic local environment where hydrophobic interaction was found to contribute most to the antigen-antibody binding probably because of the exclusion of solvent from nearby polar interactions [22].
It can be seen from Figure 4 that more aromatic residues are found in AI, more energetic deviation can be observed between the computational and experimental results under the same systemic error. If the aromatic interfacial residues in AI function separately without the cooperative effects between each other, the energetic contribution of them will be roughly additive so that the positive and linear correlation will not be observed. In summary, the rough correlation in Figure 4 suggested that there may be some unknown cooperative effects existing within the AI and these effects will increase with more and more Tyr or Trp residues stacking together. In other words, if one aromatic residue in AI is mutated into non-aromatic, the introduced effects to the binding affinity could be much larger than our expectation. This finding has gained supports from many instances [33, 34, 35]. For example, the mutation of an aromatic residue in AI of VLTrp92 (IMGT number: V-KAPPA Trp108 [36]) to Leu could lead to 1,000 fold decreases in binding affinity in the complex of D1.3/HEL [33]. In another interesting study of two antibodies to the antigen vascular endothelial growth factor (PDB code 1cz8 and 1bj1), one single mutation of VHHis101 (IMGT number: VH His109) to Tyr would cause 14 fold of affinity enhancement [34, 35].Our further analysis found this mutation will cause the AI size expanding from 4 to 7.
As to the computational methodologies to calculate the binding free energy, there have been several methods available such as MM/PBSA and other methods [37]. Although molecular dynamics methodology may give more accurate calculation [38, 39, 40, 41], these methods are avoided considering that the computing time is overwhelming for 82 antibody-antigen complexes. No matter what method is adopted, the trend for deviation between the computational and experimental results has certain reliability under the same systemic error. Similar trend was also obtained by an alternative method Rosetta (Additional file 4 - Figure S1) [42].
Conclusion
In summary, a large-scale analysis is done focusing on the spatial distribution and energetic contribution of aromatic interfacial residues for representative antibody interfaces. The results of this paper reveal the wide existence of “Aromatic Island” at the antibody interfaces where Tyr and Trp residues cluster together in an ordered way along the cleft of antigen surface. The gathering of aromatic side chains probably produces an extra cooperative effect which contributes significantly to the binding affinity between antigen and antibody. The finding of “Aromatic Island” to some extent supports the “molecular crowding” speculation in the association interface. On the other hand, the collective effects resulted from the aromatic side chains might also form the optimized local environment, which could flexibly accommodate and recognize a variety of spatial epitopes, and stabilize the molecular complexes. Future work on their exact function will not only benefit the intricate mechanism of immune recognition and specificity, but also facilitate the antibody engineering, antigen docking and potential clinical applications.
Methods
Dataset of antigen-antibody complexes
Four hundred and sixty four structures of antibody and protein antigen complexes have been obtained from the Protein Data Bank, dated Aug, 2008 and IMGT/3Dstructure-DB. In Jan, 2010, the PDB database was rescanned. 45 new crystal structures of antibody-antigen complexes were deposited and appended in our dataset. Totally, there were 509 antibody-antigen complexes included in our dataset. Those with resolution better than 3.0Å and length of antigen molecules more than 25 residues were requested to guarantee the protein antigen instead of peptide antigen. Considering that antibodies have highly similar sequence similarity, the redundancy is removed according to the spatial epitopes of protein antigen. In our study, strict criteria were set to compare the similarity of spatial epitopes between different complexes. If the identical interfacial residues between two spatial epitopes are above 20%, only one complex with the best resolution is kept. The final dataset of immune complexes was composed of 82 antibody-antigen complexes. The detailed list can be found in Additional file 2for the PDB IDs of antibody-antigen complexes included in our dataset.
Within these complexes, the interfacial residues of antibody were defined according to the change of solvent accessible surface area (ΔSASA) for each residue in unbound and bound structures [43].
ΔSASA = SASA unbound – SASA bound (1)
The value of solvent accessible surface area was calculated with program NACCESS [44]. The radius of water probe was set to be 1.4 Å [15]. The unbound structure of proteins derived directly from the structure file of complexes. The interfacial residues were those with ΔSASA more than 1Å2.
Residue connectivity at the antibody interface
The computational alanine scanning mutagenesis method was adopted to retrieve the residue contacts for the antibody interface. The interfacial residues of antibodies were mutated into Ala one by one. Comparing the SASA for every interfacial residue between mutants and wild type, the feature of spatial contact between interfacial residues of antibody was determined according to the change of SASA. Through the spatial contiguity, the distribution pattern for interfacial residues at the antibody interface was analyzed.
Random simulation of residue re-arrangement for antibody interface
To study whether the distribution pattern for clustered aromatic residues is of statistical significance, especially when the number of aromatic residues increases at the interface of antibody, the random re-arrangement of interfacial residues was made for simulative interface and compared to the actual interface of antibody. With the same solvent accessible surface area and residue composition, the interfacial residues and whole binding interface were simplified as the circles. The substituted circle for every residue was rigid, and the radius for every circle could be determined with the formula to calculate the area of circle, S = π×r 2 , where S was the area of substituted circle and equal to the SASA, and r was the radius. Then, the computational simulation was processed to simulate the pattern of random re-arrangement for interfacial residues. The substituted circles for interfacial residues were randomly laid in the big circle representing the whole interface of antibody. During the simulation, the first one circle was laid at the center. Then, the following circles were randomly laid nearest to the center one. Until all circles were laid into the big circle, a complete simulation was finished. Considering the gaps between the individual artificial circles, the boundary of the big circle is set to be flexible and exceeding of 5 Å is allowed. Every binding interface of antibody was simulated 10,000 times. The distribution of aromatic Tyr and Trp interfacial residues were investigated with comparison between the actual interfaces and the simulated ones. The statistical significance of the AI phenomenon was inspected with a test against the empirical distribution among the simulated binding interfaces. The likelihood was directly calculated among 10,000 simulations for observing the AI with the same residue composition and size as the actual interface When the probability was less than 5.0%, the AI phenomenon at the interface of antibody is considered with the statistical significance.
Calculation of binding affinity for immune complexes
In order to discuss the possible energetic contribution derived from the enriched aromatic interfacial residues, molecular modeling methods were adopted to calculate the binding affinity for immune complexes. The binding free energy was selected to evaluate the binding affinity in our research. The 3-D structure of complexes from PDB was prepared by adding hydrogen atoms in InsightII program package. Then structure was optimized by 500 steps of steepest decent followed by 1000 steps of adopted basis Newton Raphson under CHARMM force field. In the minimization process, explicit solvent molecules TIP3 were used to solvate the antibody-antigen complex. Optimized structure was saved for subsequent calculation of the binding affinity, while the water molecules were discarded. Program of DCOMPLEX was used to calculate the binding free energy [45]. On the other hand, the experimental binding affinity was derived based on the binding constant in literature, including k on , k off , K D and K A , calculated following the thermodynamic method:
ΔG = -RTln K A (4)
K A = 1/ K D = k on / k off (5)
Declarations
Acknowledgements
We thank Dr. Jie Liang and Dr. Yuzong Chen for constructive discussion. This work was supported in part by grants from Ministry of Science and Technology China (2010CB833601, 2006AA02312, 2009zx10004-601), Shanghai Municipal Education Commission (2000236018, 2000236016).
This article has been published as part of Immunome Research Volume 6 Supplement 1, 2010: Ninth International Conference on Bioinformatics (InCoB2010): Immunome Research. The full contents of the supplement are available online at http://www.immunome-research.com/supplements/6/S1.
Authors’ Affiliations
References
- Jin L, Fendly BM, Wells JA: High Resolution functional analysis of antibody-antigen interactions. J. Mol. Biol. 1992, 226: 851-865. 10.1016/0022-2836(92)90636-X.View ArticlePubMedGoogle Scholar
- Kabat EA, Wu TT, Bilofsky H: Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J. Biol. Chem. 1977, 252: 6609-6616.PubMedGoogle Scholar
- Berek C, Griffiths GM, Milstein C: Molecular events during maturation of the immune response to oxazolone. Nature. 1985, 316: 412-418. 10.1038/316412a0.View ArticlePubMedGoogle Scholar
- Wysocki L, Manser T, Gefter ML: Somatic evolution of variable region structures during an immune response. Proc. Natl. Acad. Sci. U.S.A. 1986, 83: 1847-1851. 10.1073/pnas.83.6.1847.PubMed CentralView ArticlePubMedGoogle Scholar
- Roberts S, Cheetham JC, Rees AR: Generation of an antibody with enhanced affinity and specificity for its antigen by protein engineering. Nature. 1987, 328: 731-734. 10.1038/328731a0.View ArticlePubMedGoogle Scholar
- Lea S, Stuart D: Analysis of antigenic surfaces of proteins. FASEB J. 1995, 9: 87-93.PubMedGoogle Scholar
- Roitt IM, Brostoff J, Male DK: Immunology. 1996, London: Mosby Year BookGoogle Scholar
- Mian IS, Bradwell AR, Olson AJ: Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991, 217: 133-151. 10.1016/0022-2836(91)90617-F.View ArticlePubMedGoogle Scholar
- Padlan EA: Anatomy of the antibody molecule. Mol. Immunol. 1994, 31: 169-217. 10.1016/0161-5890(94)90001-9.View ArticlePubMedGoogle Scholar
- Davies DR, Cohen GH: Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. U.S.A. 1996, 93: 7-12. 10.1073/pnas.93.1.7.PubMed CentralView ArticlePubMedGoogle Scholar
- Tsai CJ, Lin SL, Wolfson HJ, Nussinov R: Studies of protein-protein interfaces, a statistical analysis of the hydrophobic effect. Protein Science. 1997, 6 (1): 53-64. 10.1002/pro.5560060106.PubMed CentralView ArticlePubMedGoogle Scholar
- Jones S, Thornton MJ: Analysis of protein-protein interaction sites using surface patch. J. Mol. Biol. 1997, 272 (1): 121-132. 10.1006/jmbi.1997.1234.View ArticlePubMedGoogle Scholar
- Glaser F, Steinberg DM, Vakser IA, Ben-Tal N: Residue frequencies and pairing preferences at protein-protein interfaces. Proteins: Struc. Funct. 2001, 43 (2): 89-102. 10.1002/1097-0134(20010501)43:2<89::AID-PROT1021>3.0.CO;2-H.View ArticleGoogle Scholar
- Jones S, Thorn JM: Principles of protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 1996, 93 (1): 13-20. 10.1073/pnas.93.1.13.PubMed CentralView ArticlePubMedGoogle Scholar
- LoConte L, Chothia C, Janin J: The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285 (5): 2177-2198. 10.1006/jmbi.1998.2439.View ArticleGoogle Scholar
- Fellouse FA, Wiesmann C, Sidhu SS: Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. U.S.A. 2004, 101: 12467-12472. 10.1073/pnas.0401786101.PubMed CentralView ArticlePubMedGoogle Scholar
- Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS: Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J. Mol. Biol. 2006, 357: 100-114. 10.1016/j.jmb.2005.11.092.View ArticlePubMedGoogle Scholar
- Padlan EA, Abergel C, Tipper JP: Identification of specificity-determining residues in antibodies. FASEB J. 1995, 9: 133-139.PubMedGoogle Scholar
- Sundberg EJ, Urrutia M, Braden BC, Isern J, Tsuchiya D, Fields BA, Malchiodi EL, Tormo J, Schwarz FP, Mariuzza RA: Estimation of the hydrophobic effect in an antigen–antibody protein–protein interface. Biochemistry. 2000, 39: 15375-15387. 10.1021/bi000704l.View ArticlePubMedGoogle Scholar
- Li YL, Urrutia M, Smith-Gill SJ, Mariuzza RA: Dissection of binding interactions in the complex between the anti-lysozyme antibody HyHEL-63 and its antigen. Biochemistry. 2003, 42: 11-22. 10.1021/bi020589+.View ArticlePubMedGoogle Scholar
- Shiroishi M, Tsumoco K, Tanaka Y, Yokota A, Nakanishi T, Kondo H, Kumagai I: Structural consequences of mutations in interfacial Tyr residues of a protein antigen-antibody complex. J. Biol. Chem. 2007, 282: 6783-6791. 10.1074/jbc.M605197200.View ArticlePubMedGoogle Scholar
- Bogan AA, Thorn KS: Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280: 1-9. 10.1006/jmbi.1998.1843.View ArticlePubMedGoogle Scholar
- DeLano WL: Unraveling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 2002, 12: 14-20. 10.1016/S0959-440X(02)00283-X.View ArticlePubMedGoogle Scholar
- Thorn KS, Bogan AA: ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001, 17: 284-285. 10.1093/bioinformatics/17.3.284.View ArticlePubMedGoogle Scholar
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res. 2000, 28: 235-242. 10.1093/nar/28.1.235.PubMed CentralView ArticlePubMedGoogle Scholar
- Kaas Q, Ruiz M, Lefranc MP: IMGT/3Dstructure-DB and IMGT/StructurealQuery, a database and a tool for immunoglobulin, T-cell receptor and MHC structural data. Nucl. Acids. Res. 2004, 32: D208-D210. 10.1093/nar/gkh042.PubMed CentralView ArticlePubMedGoogle Scholar
- Ehrenmann F, Kaas Q, Lefranc MP: IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or antibodies, T-cell receptors, MHC, IgSF and MhcSF. Nucl. Acids. Res. 2010, 38: D301-D307. 10.1093/nar/gkp946.PubMed CentralView ArticlePubMedGoogle Scholar
- Cho KI, Lee KY, Lee KH, Kim DS, Lee DH: Specificity of molecular interactions in transient protein-protein interaction interfaces. PROTEINS: Struct. Funct. Bioinformatics. 2006, 65: 593-606. 10.1002/prot.21056.View ArticleGoogle Scholar
- Koide S, Sidhu SS: The importance of being Tyrosine: Lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chemical Biology. 2009, 4: 325-334. 10.1021/cb800314v.PubMed CentralView ArticlePubMedGoogle Scholar
- Zielenkiewicz P, Saenger W: Residue solvent accessibilities in the unfolded polypeptide chain. Biophys. J. 1992, 63: 1483-1486. 10.1016/S0006-3495(92)81746-0.PubMed CentralView ArticlePubMedGoogle Scholar
- SantaLucia J, Hicks D: The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 3: 415-440. 10.1146/annurev.biophys.32.110601.141800.View ArticleGoogle Scholar
- Rutledge LR, Campbell-Verduyn LS, Wetmore SD: Characterization of the stacking interactions between DNA or RNA nucleobases and the aromatic amino acids. Chemical Physical Letters. 2007, 444: 167-175. 10.1016/j.cplett.2007.06.090.View ArticleGoogle Scholar
- Ysern X, Fields BA, Bhat TN, Goldbaum FA, Dall’Acqua W, Schwarz FP, Poljak RJ, Mariuzza A: Solvent rearrangement in an antigen-antibody interface introduced by site-directed mutagenesis of the antibody combining site. J. Mol. Biol. 1994, 238: 496-500. 10.1006/jmbi.1994.1309.View ArticlePubMedGoogle Scholar
- Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM: VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4A resolution and mutational analysis of the interface. Structuer. 1998, 6: 1153-1167. 10.1016/S0969-2126(98)00116-6.View ArticleGoogle Scholar
- Chen Y, Wiesmann C, Fuh G, Li B, Christinger HW, McKay P, de Vos AM, Lowman HB: Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 1999, 293: 865-881. 10.1006/jmbi.1999.3192.View ArticlePubMedGoogle Scholar
- Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, Lefranc G: IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 2003, 27: 55-77. 10.1016/S0145-305X(02)00039-3.View ArticlePubMedGoogle Scholar
- Gilson MK, Zhou HX: Protein-Ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36: 21-42. 10.1146/annurev.biophys.36.040306.132550.View ArticlePubMedGoogle Scholar
- Kollman PA: Free energy calculation-applications to chemical and biochemical phenomena. Chem Rev. 1993, 93: 2395-2417. 10.1021/cr00023a004.View ArticleGoogle Scholar
- Gilson MK, Given JA, Bush BL, McCammon JA: The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J. 1997, 72: 1047-1069. 10.1016/S0006-3495(97)78756-3.PubMed CentralView ArticlePubMedGoogle Scholar
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE: Calculating structures and free eneries of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000, 33: 889-897. 10.1021/ar000033j.View ArticlePubMedGoogle Scholar
- Swanson JMJ, Henchman RH, McCammon JA: Revisiting free energy calculations: a theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys J. 2004, 86: 67-74. 10.1016/S0006-3495(04)74084-9.PubMed CentralView ArticlePubMedGoogle Scholar
- Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D: Design of a novel globular protein fold with atomic-level accuracy. Science. 2003, 302: 1364-1368. 10.1126/science.1089427.View ArticlePubMedGoogle Scholar
- Lee B, Richards FM: The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971, 55: 379-400. 10.1016/0022-2836(71)90324-X.View ArticlePubMedGoogle Scholar
- Hubbard SJ, Thornton JM: 'NACCESS', Computer Program. Department of Biochemistry and Molecular Biology, University College London. 1993Google Scholar
- Liu S, Zhang C, Zhou H, Zhou Y: A physical reference state unifies the structure-derived potential of mean force for protein folding and binding. Proteins. 2004, 56: 93-101. 10.1002/prot.20019.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.