
NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure
- Research
- Open Access
NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure
- Received: 22 September 2010
- Accepted: 13 November 2010
- Published: 13 November 2010
Abstract
Background
Binding of peptides to Major Histocompatibility class II (MHC-II) molecules play a central role in governing responses of the adaptive immune system. MHC-II molecules sample peptides from the extracellular space allowing the immune system to detect the presence of foreign microbes from this compartment. Predicting which peptides bind to an MHC-II molecule is therefore of pivotal importance for understanding the immune response and its effect on host-pathogen interactions. The experimental cost associated with characterizing the binding motif of an MHC-II molecule is significant and large efforts have therefore been placed in developing accurate computer methods capable of predicting this binding event. Prediction of peptide binding to MHC-II is complicated by the open binding cleft of the MHC-II molecule, allowing binding of peptides extending out of the binding groove. Moreover, the genes encoding the MHC molecules are immensely diverse leading to a large set of different MHC molecules each potentially binding a unique set of peptides. Characterizing each MHC-II molecule using peptide-screening binding assays is hence not a viable option.
Results
Here, we present an MHC-II binding prediction algorithm aiming at dealing with these challenges. The method is a pan-specific version of the earlier published allele-specific NN-align algorithm and does not require any pre-alignment of the input data. This allows the method to benefit also from information from alleles covered by limited binding data. The method is evaluated on a large and diverse set of benchmark data, and is shown to significantly out-perform state-of-the-art MHC-II prediction methods. In particular, the method is found to boost the performance for alleles characterized by limited binding data where conventional allele-specific methods tend to achieve poor prediction accuracy.
Conclusions
The method thus shows great potential for efficient boosting the accuracy of MHC-II binding prediction, as accurate predictions can be obtained for novel alleles at highly reduced experimental costs. Pan-specific binding predictions can be obtained for all alleles with know protein sequence and the method can benefit by including data in the training from alleles even where only few binders are known. The method and benchmark data are available at https://services.healthtech.dtu.dk
Keywords
- Predictive Performance
- Binding Data
- Peptide Core
- Binding Prediction
- Peptide Binding Affinity
Background
Binding of peptides to MHC II molecules play a major role in governing adaptive immune responses. They allow peptides derived from pathogens in the extracellular compartment to be presented by professional antigen presenting cells (APCs) to T helper cells of the immune system.
These T cells might in turn activate the presenting cell to kill intracellular bacterial infections. Help is also for most antigens needed to activate B cells to produce antibodies that may neutralize the pathogen. Over the last decade a number of different methods for prediction of binding to MHC II molecules have been developed, the most known being the TEPITOPE method [1]. Prediction of binding of peptides to MHC II is complicated by the immense polymorphism of the MHC class II alleles since the many different encoded MHC class II molecules (more than 690 different known HLA-DR alleles are known) bind very different sets of peptides. The TEPITOPE method covers 50 of these HLA-DR alleles. During the last decays several data driven so-called allele-specific methods have been developed for alleles where sufficient numbers of binding peptides are known. These methods cover a very broad range of different bioinformatics training algorithms including Gibbs sampler [2, 3], artificial neural networks [4, 5], support vector machines [6, 7, 8], hidden Markov models [9], as well as other (often exotic) motif search algorithms [10, 11, 12, 13, 14, 15, 16, 17, 18]. For a detailed review please refer to Nielsen et al. [19].
These methods can interpolate between peptide binding data and create predictions for peptides not present in the training set. Recently, pan-specific methods that in principle can make predictions for all alleles with known amino acid sequence have been developed [20, 21, 22, 23, 24, 25, 26]. These methods work by including information about the amino acid sequence of the MHC molecule as input to the method allowing the methods to integrate information across multiple alleles simultaneously thus boosting the predictive performance and potentially extrapolate the predictions to previously un-characterized MHC molecules. Several benchmark calculations have demonstrated the power of such pan-specific methods [27] and have shown how accurate predictions can be obtained also for alleles for which no or very limited binding data have been identified [21, 28].
One of the best performing pan-specific MHC class II prediction method is the NetMHCIIpan method [29]. An important limiting factor for this method lies in the need for a pre-alignment of the input training data identifying the peptide-binding core prior to the training of the method. Such pre-alignments require sufficient data being available for all MHC molecules included in the training data in order to derive accurate allele-specific predictions. It has earlier been shown that this number of peptide binding data for MHC class II is of the order of many hundred [3, 19], which makes it very costly to develop accurate MHC class II predictions. In order to circumvent this, we here propose a less demanding, yet highly efficient method to generate MHC class II predictors. This method is a pan-specific version of the earlier published allele-specific NN-align algorithm [5] and does not require any pre-alignment of the input data. The method hence has the potential to benefit also from information from alleles covered by limited binding data. Here, we demonstrate its predictive power in a series of large-scale benchmark calculations.
Materials and methods
Data
Quantitative HLA-DR peptide binding data
|
Allele |
# |
# bind |
Allele |
# |
# bind |
|---|---|---|---|---|---|
|
DRB1*0101 |
7685 |
4382 |
DRB1*1101 |
1794 |
778 |
|
DRB1*0301 |
2505 |
649 |
DRB1*1201 |
117 |
81 |
|
DRB1*0302 |
148 |
44 |
DRB1*1202 |
117 |
79 |
|
DRB1*0401 |
3116 |
1039 |
DRB1*1302 |
1580 |
493 |
|
DRB1*0404 |
577 |
336 |
DRB1*1402 |
118 |
78 |
|
DRB1*0405 |
1582 |
627 |
DRB1*1404 |
30 |
16 |
|
DRB1*0701 |
1745 |
849 |
DRB1*1412 |
116 |
63 |
|
DRB1*0802 |
1520 |
431 |
DRB1*1501 |
1769 |
709 |
|
DRB1*0806 |
118 |
91 |
DRB3*0101 |
1501 |
281 |
|
DRB1*0813 |
1370 |
455 |
DRB3*0301 |
160 |
70 |
|
DRB1*0819 |
116 |
54 |
DRB4*0101 |
1521 |
485 |
|
DRB1*0901 |
1520 |
622 |
DRB5*0101 |
3106 |
1280 |
|
Total |
33931 |
13992 |
MHC class II ligands from the SYFPEITHI database
|
Allele |
# |
Allele |
# |
|---|---|---|---|
|
HLA-DRB1*0101 |
53 |
HLA-DRB1*1101 |
35 |
|
HLA-DRB1*0102 |
5 |
HLA-DRB1*1104 |
8 |
|
HLA-DRB1*0301 |
88 |
HLA-DRB1*1201 |
11 |
|
HLA-DRB1*0401 |
468 |
HLA-DRB1*1301 |
16 |
|
HLA-DRB1*0402 |
36 |
HLA-DRB1*1302 |
19 |
|
HLA-DRB1*0403 |
1 |
HLA-DRB1*1401 |
9 |
|
HLA-DRB1*0404 |
42 |
HLA-DRB1*1501 |
22 |
|
HLA-DRB1*0405 |
36 |
HLA-DRB1*1502 |
3 |
|
HLA-DRB1*0701 |
47 |
HLA-DRB1*1601 |
2 |
|
HLA-DRB1*0801 |
39 |
HLA-DRB3*0101 |
2 |
|
HLA-DRB1*0802 |
1 |
HLA-DRB3*0301 |
5 |
|
HLA-DRB1*0803 |
1 |
HLA-DRB4*0101 |
6 |
|
HLA-DRB1*0901 |
6 |
HLA-DRB4*0103 |
2 |
|
HLA-DRB1*1001 |
183 |
HLA-DRB5*0101 |
18 |
|
Total |
1164 |
HLA-DR restriction T cell epitope from the IEDB database
|
Allele |
# |
Allele |
# |
|---|---|---|---|
|
HLA-DRB1*0101 |
125 |
HLA-DRB1*1103 |
3 |
|
HLA-DRB1*0102 |
4 |
HLA-DRB1*1104 |
6 |
|
HLA-DRB1*0103 |
5 |
HLA-DRB1*1201 |
3 |
|
HLA-DRB1*0301 |
173 |
HLA-DRB1*1301 |
15 |
|
HLA-DRB1*0401 |
342 |
HLA-DRB1*1302 |
10 |
|
HLA-DRB1*0402 |
33 |
HLA-DRB1*1303 |
3 |
|
HLA-DRB1*0403 |
14 |
HLA-DRB1*1401 |
16 |
|
HLA-DRB1*0404 |
46 |
HLA-DRB1*1404 |
1 |
|
HLA-DRB1*0405 |
21 |
HLA-DRB1*1405 |
2 |
|
HLA-DRB1*0406 |
6 |
HLA-DRB1*1501 |
193 |
|
HLA-DRB1*0407 |
4 |
HLA-DRB1*1502 |
20 |
|
HLA-DRB1*0408 |
2 |
HLA-DRB1*1503 |
2 |
|
HLA-DRB1*0701 |
56 |
HLA-DRB1*1601 |
5 |
|
HLA-DRB1*0703 |
1 |
HLA-DRB1*1602 |
3 |
|
HLA-DRB1*0801 |
4 |
HLA-DRB3*0101 |
12 |
|
HLA-DRB1*0802 |
2 |
HLA-DRB3*0202 |
10 |
|
HLA-DRB1*0803 |
2 |
HLA-DRB3*0301 |
1 |
|
HLA-DRB1*0901 |
13 |
HLA-DRB4*0101 |
17 |
|
HLA-DRB1*1001 |
4 |
HLA-DRB4*0103 |
1 |
|
HLA-DRB1*1101 |
88 |
HLA-DRB5*0101 |
55 |
|
HLA-DRB1*1102 |
1 |
HLA-DRB5*0102 |
1 |
|
Total |
1325 |
Method
The pan-specific NetMHCIIpan-2.0 method is a hybrid of the earlier published methods for pan-specific for MHC class I and class II binding, NetMHCpan [20, 21], and NetMHCIIpan [29], and the NN-align method recently published for allele-specific MHC class II binding predictions [5]. The overall method architecture is similar to the NN-align method, and the manner in which the MHC polymorphism method is incorporated is similar to that of the NetMHCpan and NetMHCIIpan methods.
The method was implemented as a conventional feed-forward artificial neural network. Like the NN-align method, the method consists of a two-step procedure that simultaneously estimates the optimal peptide binding register (core) and network weight configuration. Initially, all network weights were assigned random values. Given this set of network weights, the core of a given peptide was identified as the highest scoring of all nonamers contained within the peptide. The score of a nonamer peptide was calculated using the conventional feed-forward algorithm. The network weights were updated using gradient descent back-propagation. Given a peptide core alignment, the weights were updated to lower the sum of squared errors between the predicted binding score and the measured binding affinity target value. A peptide core was presented to the network as described for the NN-align method including encoding of peptide flanking regions (PFR), PFR length and the peptide length. The MHC environment defining the peptide binding strength was implemented in terms of the MHC pseudo sequence constructed from 21 polymorphic amino acid positions in potential contact with the bound peptide as described by Nielsen et al. [29]. Two types of sequence encodings (sparse and blosum) were applied for the peptide-core and MHC pseudo sequences as described by Nielsen et al. [32]. For each peptide core, the input to the neural network thus consisted of the peptide core and MHC environment residues ((9+21) × 20 = 600 inputs), the PFRs (2 × 20 = 40 inputs), the peptide length (2 inputs), the length of the C and N terminal PFR's (2 × 2 = 4 inputs) resulting in a total of 646 input values. The peptide binding affinity IC50 values were encoded to the neural network as log-transformed values, using the relation 1-log (aff)/log(15,000), where aff is the measured binding affinity (IC50) in nM units [32]. Note, that we here use 15,000 as the base for the logarithmic transformation. This is in contrast to the 50,000 used in previous works by our group, and is chosen due to a lower sensitivity for weak binding peptides of the high-throughput peptide-binding screening assay described by Justesen et al. [30].
The networks were trained using 5-fold cross-validation. Network ensembles were trained with 40 hidden neurons. The procedure of i) identifying the optimal peptide core, and ii) updating the network weights to lower the predictive error was repeated for 500 cycles. Since the "search landscape" has a large set of local minima each with close to identical performance values, the network training was repeated 10 times, each with different initial configuration values. This led to significantly improved prediction accuracy (data not shown). In total 20 (2 encoding schemes*10 seeds) networks were created for each training/test set configuration. The binding core of a given peptide was assigned by a majority vote of the networks in the ensemble.
Leave-one-out (LOO) network training and benchmark
Leave-one-(allele)-out experiments were conducted to investigate the predictive performance of the method in situations where binding data for a given allele was excluded from the training. Two types of LOO experiments were conducted. In the first type, peptide-binding data for a given allele were excluded from the training of the prediction method, and upon training, the predictive performance was evaluated using the peptide binding affinity values for the HLA-DR molecule in question. This is the LOO approach applied in the NetMHCIIpan-1.0 method [29] and allows for a direct comparison of this method to the method proposed here when trained on similar data sets.
Since many of the peptides in the training data have been measured for binding affinities to multiple alleles, the above LOO experiment can lead to a significant overestimation of the performance for a given prediction method. To reduce this bias, a second type of LOO experiment was conducted where all data representing a given peptide was excluded from the training of the prediction method. That is, if a given peptide was measured against multiple alleles including the allele in question, all these measurements were excluded from the LOO training. To avoid reducing the size of the training data too much, this second type of LOO training was performed as a three-fold cross-validation for alleles characterized by more than 200 data points.
Performance measures
The predictive performance was measured in terms of the area under the ROC curve (AUC) value and Pearson's correlation coefficient (PCC). The receiver operating characteristic (ROC) curve is a graphical plot of the sensitivity versus the false positive rate (1 - specificity) as the discrimination threshold is varied. Through out this work, a binding threshold value of 500 nM was used to classify the peptides. The area under the ROC curve (AUC) gives an indication of the accuracy of a prediction method. An AUC value of 1 corresponds to perfect predictions and a value of 0.5 reflects random predictions. Likewise, PCC is a measure of the accuracy of a prediction method. It is obtained by dividing the covariance of the two variables by the product of their standard deviations. For perfect predictions PCC is 1 (or -1), and for random predictions PCC is 0.
Nearest neighbor distance calculation
The distance between two MHC alleles was estimated as described by Nielsen et al. [29] using the relation 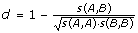 , where s(A, B) is the BLOSUM50 similarity score between the pseudo sequences of allele A and B, respectively. Next, the nearest neighbor distance for an allele is defined as the minimal distance to any allele included in the training data set.
, where s(A, B) is the BLOSUM50 similarity score between the pseudo sequences of allele A and B, respectively. Next, the nearest neighbor distance for an allele is defined as the minimal distance to any allele included in the training data set.
Results
Pan-specific versus allele-specific predictions
Five-fold cross-validation performance of the pan-specific NetMHCIIpan-2.0 method compared to the allele-specific NN-align and TEPITOPE methods on the quantitative benchmark data set
|
NN-align |
NetMHCIIpan-2.0 |
TEPITOPE |
|||||
|---|---|---|---|---|---|---|---|
|
Allele |
# |
# bind |
PCC |
AUC |
PCC |
AUC |
AUC |
|
DRB1*0101 |
7685 |
4382 |
0.675 |
0.825 |
0.711 |
0.846 |
0.727 |
|
DRB1*0301 |
2505 |
649 |
0.690 |
0.855 |
0.709 |
0.864 |
0.718 |
|
DRB1*0302 |
148 |
44 |
0.272 |
0.659 |
0.525 |
0.757 |
|
|
DRB1*0401 |
3116 |
1039 |
0.643 |
0.833 |
0.670 |
0.848 |
0.762 |
|
DRB1*0404 |
577 |
336 |
0.565 |
0.766 |
0.630 |
0.818 |
0.747 |
|
DRB1*0405 |
1582 |
627 |
0.710 |
0.869 |
0.698 |
0.858 |
0.780 |
|
DRB1*0701 |
1745 |
849 |
0.718 |
0.855 |
0.740 |
0.864 |
0.777 |
|
DRB1*0802 |
1520 |
431 |
0.518 |
0.778 |
0.526 |
0.780 |
0.645 |
|
DRB1*0806 |
118 |
91 |
0.744 |
0.902 |
0.796 |
0.924 |
0.884 |
|
DRB1*0813 |
1370 |
455 |
0.729 |
0.878 |
0.746 |
0.885 |
0.750 |
|
DRB1*0819 |
116 |
54 |
0.370 |
0.706 |
0.608 |
0.808 |
|
|
DRB1*0901 |
1520 |
622 |
0.597 |
0.810 |
0.634 |
0.818 |
|
|
DRB1*1101 |
1794 |
778 |
0.756 |
0.873 |
0.777 |
0.883 |
0.793 |
|
DRB1*1201 |
117 |
81 |
0.699 |
0.860 |
0.764 |
0.892 |
|
|
DRB1*1202 |
117 |
79 |
0.695 |
0.866 |
0.769 |
0.900 |
|
|
DRB1*1302 |
1580 |
493 |
0.630 |
0.819 |
0.634 |
0.825 |
0.596 |
|
DRB1*1402 |
118 |
78 |
0.623 |
0.825 |
0.694 |
0.860 |
|
|
DRB1*1404 |
30 |
16 |
0.466 |
0.661 |
0.613 |
0.737 |
|
|
DRB1*1412 |
116 |
63 |
0.680 |
0.857 |
0.757 |
0.894 |
|
|
DRB1*1501 |
1769 |
709 |
0.641 |
0.815 |
0.653 |
0.819 |
0.731 |
|
DRB3*0101 |
1501 |
281 |
0.673 |
0.843 |
0.690 |
0.850 |
|
|
DRB3*0301 |
160 |
70 |
0.604 |
0.826 |
0.736 |
0.853 |
|
|
DRB4*0101 |
1521 |
485 |
0.701 |
0.854 |
0.675 |
0.837 |
|
|
DRB5*0101 |
3106 |
1280 |
0.735 |
0.865 |
0.765 |
0.882 |
0.760 |
|
Ave |
33931 |
13992 |
0.631 |
0.821 |
0.688 |
0.846 |
|
|
Ave* |
0.673 |
0.841 |
0.697 |
0.854 |
0.744 |
||
|
Ave** |
0.580 |
0.797 |
0.679 |
0.837 |
|||
From the results in table 4, it is apparent that the NetMHCIIpan-2.0 method significantly outperforms both the NN-align and TEPITOPE methods (p < 0.01, binomial test). For 9 of the 9 alleles covered by less than 400 peptide-binding measurements, we find that NetMHCIIpan-2.0 outperforms NN-align. These results strongly indicate that NetMHCIIpan-2.0 is capable of benefiting from information from the multiple alleles included in the benchmark to boost the predictive performance and deliver accurate predictions also for alleles covered by limited binding data. Only for 3 out of the 24 alleles does the NN-align perform better than NetMHCIIpan-2.0. These alleles are all covered by more than 1500 peptide-binding measurements. This hence confirms the results obtained earlier for MHC class I binding predictions namely that pan-specific predictions are particularly beneficial when binding data are scarce or absent [28]. What is also clear from the data in table 4 is that the NetMHCIIpan-2.0 method is capable of maintaining its high performance also for alleles not characterized by the TEPITIOPE method.
NetMHCpanII-1.0 versus NetMHCpanII-2.0
LOO benchmark comparison of the pan-specific NetMHCIIpan-2.0 and the NetMHCIIpan-1.0 methods
|
NetMHCIIpan-1.0 |
NetMHCIIpan-2.0 |
TEPITOPE |
|||||
|---|---|---|---|---|---|---|---|
|
Allele |
# |
#bind |
PCC |
AUC |
PCC |
AUC |
AUC |
|
DRB1*0101 |
5166 |
3510 |
0.571 |
0.778 |
0.627 |
0.794 |
0.720 |
|
DRB1*0301 |
1020 |
277 |
0.465 |
0.746 |
0.560 |
0.792 |
0.664 |
|
DRB1*0401 |
1024 |
510 |
0.591 |
0.775 |
0.652 |
0.802 |
0.716 |
|
DRB1*0404 |
663 |
386 |
0.693 |
0.852 |
0.731 |
0.869 |
0.770 |
|
DRB1*0405 |
630 |
425 |
0.594 |
0.808 |
0.626 |
0.823 |
0.759 |
|
DRB1*0701 |
853 |
498 |
0.655 |
0.825 |
0.753 |
0.886 |
0.761 |
|
DRB1*0802 |
420 |
148 |
0.637 |
0.841 |
0.700 |
0.869 |
0.766 |
|
DRB1*0901 |
530 |
254 |
0.406 |
0.653 |
0.474 |
0.684 |
|
|
DRB1*1101 |
950 |
429 |
0.580 |
0.799 |
0.721 |
0.875 |
0.721 |
|
DRB1*1302 |
498 |
199 |
0.323 |
0.658 |
0.337 |
0.648 |
0.652 |
|
DRB1*1501 |
934 |
450 |
0.533 |
0.738 |
0.598 |
0.769 |
0.686 |
|
DRB3*0101 |
549 |
75 |
0.449 |
0.716 |
0.474 |
0.733 |
|
|
DRB4*0101 |
446 |
200 |
0.448 |
0.724 |
0.515 |
0.762 |
|
|
DRB5*0101 |
924 |
478 |
0.627 |
0.831 |
0.722 |
0.879 |
0.686 |
|
Ave |
0.541 |
0.768 |
0.606 |
0.799 |
|||
|
Ave* |
0.570 |
0.786 |
0.639 |
0.819 |
0.718 |
||
The extended LOO benchmark
|
OLD |
NEW |
TEPITOPE |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Allele |
# |
#bind |
PCC |
AUC |
NN |
dist |
PCC |
AUC |
NN |
dist |
AUC |
|
DRB1*0101 |
7685 |
4382 |
0.567 |
0.767 |
DRB1*0401 |
0.352 |
0.583 |
0.786 |
DRB1*1402 |
0.322 |
0.727 |
|
DRB1*0301 |
2505 |
649 |
0.433 |
0.727 |
DRB3*0101 |
0.277 |
0.499 |
0.765 |
DRB1*0302 |
0.156 |
0.718 |
|
DRB1*0401 |
3116 |
1039 |
0.563 |
0.787 |
DRB1*0405 |
0.066 |
0.594 |
0.804 |
DRB1*0405 |
0.066 |
0.762 |
|
DRB1*0404 |
577 |
336 |
0.592 |
0.806 |
DRB1*0401 |
0.091 |
0.595 |
0.804 |
DRB1*0401 |
0.091 |
0.747 |
|
DRB1*0405 |
1582 |
627 |
0.638 |
0.826 |
DRB1*0401 |
0.066 |
0.633 |
0.833 |
DRB1*0401 |
0.066 |
|
|
DRB1*0701 |
1745 |
849 |
0.659 |
0.831 |
DRB1*0901 |
0.504 |
0.648 |
0.826 |
DRB1*0901 |
0.504 |
0.780 |
|
DRB1*0802 |
1520 |
431 |
0.380 |
0.710 |
DRB1*1101 |
0.111 |
0.369 |
0.692 |
DRB1*0813 |
0.041 |
0.777 |
|
DRB1*0901 |
1520 |
622 |
0.539 |
0.757 |
DRB5*0101 |
0.431 |
0.517 |
0.762 |
DRB5*0101 |
0.431 |
0.645 |
|
DRB1*1101 |
1794 |
778 |
0.602 |
0.799 |
DRB1*1302 |
0.084 |
0.460 |
0.741 |
DRB1*1302 |
0.084 |
|
|
DRB1*1302 |
1580 |
493 |
0.338 |
0.691 |
DRB1*1101 |
0.084 |
0.323 |
0.671 |
DRB1*1101 |
0.084 |
0.793 |
|
DRB1*1501 |
1769 |
709 |
0.568 |
0.775 |
DRB1*0404 |
0.295 |
0.525 |
0.756 |
DRB1*0404 |
0.295 |
0.596 |
|
DRB3*0101 |
1501 |
281 |
0.339 |
0.672 |
DRB1*0301 |
0.277 |
0.374 |
0.702 |
DRB3*0301 |
0.223 |
0.731 |
|
DRB4*0101 |
1521 |
485 |
0.506 |
0.753 |
DRB1*0404 |
0.397 |
0.518 |
0.766 |
DRB1*0404 |
0.397 |
|
|
DRB5*0101 |
3106 |
1280 |
0.547 |
0.781 |
DRB1*1101 |
0.295 |
0.608 |
0.813 |
DRB1*1101 |
0.295 |
|
|
DRB1*0302 |
148 |
44 |
0.396 |
0.729 |
DRB1*0301 |
0.156 |
0.542 |
0.759 |
DRB1*1402 |
0.119 |
0.760 |
|
DRB1*0806 |
118 |
91 |
0.670 |
0.886 |
DRB1*0802 |
0.107 |
0.703 |
0.902 |
DRB1*0802 |
0.107 |
|
|
DRB1*0813 |
1370 |
455 |
0.505 |
0.735 |
DRB1*0802 |
0.041 |
0.340 |
0.666 |
DRB1*0802 |
0.041 |
0.884 |
|
DRB1*0819 |
116 |
54 |
0.567 |
0.789 |
DRB1*0802 |
0.107 |
0.566 |
0.813 |
DRB1*0813 |
0.083 |
0.750 |
|
DRB1*1201 |
117 |
81 |
0.626 |
0.786 |
DRB1*1101 |
0.445 |
0.609 |
0.798 |
DRB1*1202 |
0.045 |
|
|
DRB1*1202 |
117 |
79 |
0.623 |
0.814 |
DRB1*1101 |
0.399 |
0.713 |
0.879 |
DRB1*1201 |
0.045 |
|
|
DRB1*1402 |
118 |
78 |
0.570 |
0.793 |
DRB1*1101 |
0.148 |
0.659 |
0.846 |
DRB1*0302 |
0.119 |
|
|
DRB1*1404 |
30 |
16 |
0.393 |
0.594 |
DRB1*0404 |
0.311 |
0.646 |
0.679 |
DRB1*0806 |
0.240 |
|
|
DRB1*1412 |
116 |
63 |
0.640 |
0.845 |
DRB1*0802 |
0.180 |
0.738 |
0.897 |
DRB1*0813 |
0.139 |
|
|
DRB3*0301 |
160 |
70 |
0.395 |
0.738 |
DRB3*0101 |
0.223 |
0.545 |
0.765 |
DRB3*0101 |
0.223 |
|
|
Ave |
0.527 |
0.766 |
0.554 |
0.780 |
|||||||
|
Ave* |
0.543 |
0.779 |
0.529 |
0.774 |
0.744 |
||||||
|
Ave** |
0.539 |
0.771 |
0.606 |
0.800 |
|||||||
The results presented in table 6 clearly demonstrate that the enrichment of novel peptide binding data with a broader allelic coverage leads to an improved predictive performance of the pan-specific prediction method. We can quantify to what degree an allele will benefit from other alleles being present in the training data by calculating its distance to the nearest neighbor in the training data (see Materials and methods). Earlier work has demonstrated that this distance measure correlates strongly with the performance of pan-specific prediction methods [20, 21]. For 10 of the 11 alleles, where including the novel 10 alleles has decreased the nearest neighbor distance, the NEW method has a higher AUC predictive performance compared to the OLD. For the remaining 13 alleles, the 10 novel alleles have not altered the nearest neighbor distance and the performance of the two methods is similar. This strongly underlines the essential prerequisite for accurate pan-specific prediction methods demonstrated earlier for MHC class I, namely a population of the close neighborhood of un-characterized MHC molecules [20, 28].
Lin benchmark
Predictive performance in terms of the AUC on the Lin benchmark data set
|
Allele |
NetMHCIIpan-2.0 |
NetMHCIIpan-1.0 |
TEPITOPE |
Multipred_SVM |
SVMHC |
|---|---|---|---|---|---|
|
DRB1*0101 |
0.883 |
0.847 |
0.919 |
0.860 |
0.860 |
|
DRB1*0301 |
0.716 |
0.668 |
0.718 |
0.800 |
0.690 |
|
DRB1*0401 |
0.846 |
0.815 |
0.745 |
0.650 |
0.750 |
|
DRB1*0701 |
0.878 |
0.852 |
0.715 |
0.700 |
0.740 |
|
DRB1*1101 |
0.884 |
0.821 |
0.824 |
0.780 |
0.830 |
|
DRB1*1301 |
0.729 |
0.715 |
0.718 |
0.630 |
0.720 |
|
DRB1*1501 |
0.838 |
0.791 |
0.737 |
0.620 |
0.660 |
|
Ave |
0.825 |
0.787 |
0.768 |
0.720 |
0.750 |
The results in table 7 clearly show that NetMHCIIpan-2.0 outperforms both the earlier NetMHCIIpan-1.0 method, as well as the other methods included in the benchmark.
Identifying MHC class II ligands and T cell epitopes
Endogenous HLA-DR ligand benchmark
|
SYF |
# |
NetMHCIIpan-1.0 |
NetMHCIIpan-2.0 |
TEPITOPE |
|---|---|---|---|---|
|
Ave per ligand |
1164 |
0.800 |
0.829 |
|
|
Ave per allele |
28 |
0.788 |
0.797 |
|
|
In TEPITOPE |
17 |
0.768 |
0.786 |
0.799 |
|
!In TEPITOPE |
11 |
0.819 |
0.814 |
|
|
IEDB |
# |
NetMHCIIpan-1.0 |
NetMHCIIpan-2.0 |
TEPITOPE |
|
Ave per epitope |
1325 |
0.729 |
0.751 |
|
|
Ave per allele |
42 |
0.759 |
0.781 |
|
|
In TEPITOPE |
20 |
0.745 |
0.747 |
0.755 |
|
!In TEPITOPE |
22 |
0.772 |
0.812 |
Both the SYFPEITI ligand and IEDB epitope benchmarks show that the NetMHCIIpan-2.0 method performs better than the original NetMHCIIpan-1.0 method. On the per ligand basis, the NetMHCIIpan-2.0 method significantly outperforms NetMHCIIpan-1.0 for both data sets (p < 0.01, binomial test excluding ties). In terms of the per-allele performance, the NetMHCIIpan-2.0 also achieved a higher performance than the NetMHCIIpan-1.0 method. This difference is however not statistically significant (p > 0.1 binomial test, in both cases). For the alleles characterized by the TEPITOPE method, the TEPITOPE method achieves the highest performance of the three methods for both data sets. This difference, however, is not statistically significant (p > 0.5 in all cases, binomial test). For alleles not characterized by TEPITOPE, the NetMHCIIpan-2.0 method significantly outperform NetMHCIIpan-1.0 for the IEDB data set (p < 0.01, binomial test), whereas the two methods for this set of alleles achieve a similar predictive performance when evaluated on the SYFPEITHI dataset.
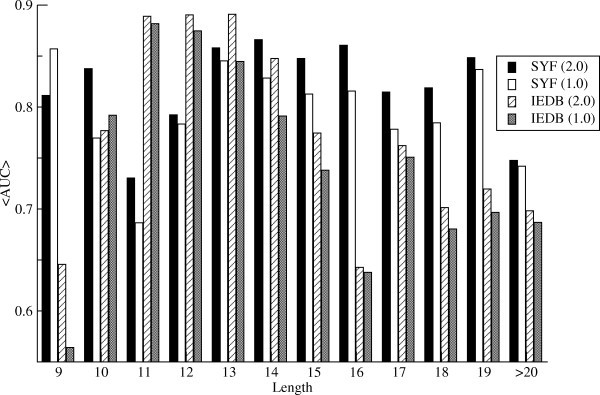
Histogram of the predictive performance measured in terms of the AUC value for the ligands/epitopes in the SYFPEITHI/IEDB dataset as a function of the peptide length. 2.0 refers to the pan-specific method developed here, and 1.0 refers to the NetMHCIIpan-1.0 method. SYF refers to the SYFPEITHI ligand data set, and IEDB refers to the IEDB T cell epitope data set.
Figure 1 clearly demonstrates that the NetMHCIIpan-2.0 method, for the majority of peptide lengths, outperforms the NetMHCIIpan-1.0 method. Only for very short peptides (length equal to 9 for the SYFPEITHI data set and length equal to 10 for the IEDB data set) does the NetMHCIIpan-1.0 achieve the highest AUC value. What is also clear for the IEDB data set is that both methods achieve their highest predictive performance for peptides of length less than 15 amino acids. The average AUC for epitopes with a length less then 15 amino acids is 0.823. This values is significantly higher than the average AUC for epitopes with a length greater than 15 (0.704, p < 0.005, t-test). This difference is not observed for the SYFPEITHI ligand data set, hence strongly suggesting that the longer epitopes in the IEDB data set are not "true" epitopes in the sense of defining the minimal HLA restriction element.
Discussion
Development of accurate prediction algorithms for MHC class II binding is complicated by the fact that the MHC class II molecule has an open binding cleft, and that peptide binders are accommodated in the binding cleft in a binding register that a priori is unknown. Training of methods for prediction of peptide-MHC class II binding hence rely on either a two step procedure where first the binding register is identified and next the aligned peptides are used to train the binding prediction algorithm or a procedure where these two steps are integrated and performed simultaneously.
We have earlier shown that developing allele-specific prediction methods for MHC class II binding using the latter approach leads to higher prediction accuracy [3, 5]. We have further for MHC class I demonstrated that training the predictors in a pan-specific manner, incorporating all binding data across multiple MHC molecules simultaneously in the training, leads to a significant boost in the predictive performance in particular for MHC molecules characterized by few or no binding data [20, 21, 22, 28].
Based on these findings, we have in this paper developed a pan-specific method for prediction of MHC class II binding affinities. The method was trained on binding data covering multiple MHC class II simultaneously, and does not require any prior alignment or binding register-identification. The method was evaluated in several large-scale benchmarks and shown consistently to outperform all other methods investigated, including state-of the-art allele-specific (NN-align [5]) and pan-specific (NetMHCIIpan [29]) methods, as well as and the well-known TEPITOPE method [1]. In particular, it was demonstrated that the proposed method due to its pan-specific nature could boost performance for alleles characterized by limited binding data, and in such cases significantly out-perform allele specific methods. The method thus demonstrates great potential for efficient boosting of the accuracy of MHC class II binding prediction, as accurate predictions can be achieved for novel alleles at a highly reduced experimental cost, and pan-specific binding predictions can be obtained for all alleles with known protein sequence by a method trained using data with limited allelic coverage.
When benchmarked on large data sets of know HLA-DR ligands and epitopes, the method was shown to have a predictive performance comparable to that of TEPITOPE for alleles covered by this method, and maybe more important maintain this high performance also for alleles not described by the TEPITOPE method.
For MHC class I, we have earlier demonstrated that a pan-specific predictor can benefit from being trained on cross-loci (and cross-species) peptide binding data [20]. The development of a cross-loci model for HLA class II is complicated by the fact that the HLA-DRA molecule is close to monomorphic (only two allelic version exists). This is in contrast to HLA-DP and HLA-DQ where both the α and β chains are highly polymorphic. Moreover, the structures of the HLA molecules are less conserved across the three loci for class II compared to class I, and finally very limited peptide binding data have been generated characterizing the different HLA-DP and DQ molecules. As of September 2010, only five HLA-DP and six HLA-DQ alleles have been characterized in the IEDB database with more than 200 peptide-binding measurements [31]. Nonetheless, large amounts of peptide binding data for the HLA-DP and HLA-DQ loci will most likely become available in the near future providing a broader allelic coverage, and future evaluations will demonstrate if also MHC class II binding prediction algorithms using training algorithms like the one outlined in this work, will benefit from pan-specific training across the different loci.
The method and benchmark data sets described in this work are available at https://services.healthtech.dtu.dk (method) and https://www.healthtech.dtu.dk (benchmark data).
Declarations
Acknowledgements
This work was supported by a NIH (National Institute of Health) grant (contract no. HHSN272200900045C) and EU 7th framework grant 222773 (Pep-ChipOmics).
Authors’ Affiliations
References
- Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, Braxenthaler M, Gallazzi F, Protti MP, Sinigaglia F, Hammer J: Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 1999, 17:555–561.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O: Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 2004, 20:1388–1397.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Lund O: Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC bioinformatics 2007, 8:238.View ArticlePubMedGoogle Scholar
- Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L: Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics 1998, 14:121–130.View ArticlePubMedGoogle Scholar
- Nielsen M, Lund O: NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC bioinformatics 2009, 10:296.View ArticlePubMedGoogle Scholar
- Wan J, Liu W, Xu Q, Ren Y, Flower DR, Li T: SVRMHC prediction server for MHC-binding peptides. BMC bioinformatics 2006, 7:463.View ArticlePubMedGoogle Scholar
- Salomon J, Flower DR: Predicting Class II MHC-Peptide binding: a kernel based approach using similarity scores. BMC bioinformatics 2006, 7:501.View ArticlePubMedGoogle Scholar
- Cui J, Han LY, Lin HH, Zhang HL, Tang ZQ, Zheng CJ, Cao ZW, Chen YZ: Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. Molecular immunology 2007, 44:866–877.View ArticlePubMedGoogle Scholar
- Noguchi H, Kato R, Hanai T, Matsubara Y, Honda H, Brusic V, Kobayashi T: Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J Biosci Bioeng 2002, 94:264–270.PubMedGoogle Scholar
- Bui HH, Sidney J, Peters B, Sathiamurthy M, Sinichi A, Purton KA, Mothe BR, Chisari FV, Watkins DI, Sette A: Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics 2005, 57:304–314.View ArticlePubMedGoogle Scholar
- Murugan N, Dai Y: Prediction of MHC class II binding peptides based on an iterative learning model. Immunome research 2005, 1:6.View ArticlePubMedGoogle Scholar
- Chang ST, Ghosh D, Kirschner DE, Linderman JJ: Peptide length-based prediction of peptide-MHC class II binding. Bioinformatics 2006, 22:2761–2767.View ArticlePubMedGoogle Scholar
- Doytchinova IA, Flower DR: Towards the in silico identification of class II restricted T-cell epitopes: a partial least squares iterative self-consistent algorithm for affinity prediction. Bioinformatics 2003, 19:2263–2270.View ArticlePubMedGoogle Scholar
- Rajapakse M, Schmidt B, Feng L, Brusic V: Predicting peptides binding to MHC class II molecules using multi-objective evolutionary algorithms. BMC bioinformatics 2007, 8:459.View ArticlePubMedGoogle Scholar
- Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S: SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999, 50:213–219.View ArticlePubMedGoogle Scholar
- Hertz T, Yanover C: PepDist: a new framework for protein-peptide binding prediction based on learning peptide distance functions. BMC bioinformatics 2006,7(Suppl 1):S3.View ArticlePubMedGoogle Scholar
- Zhang W, Liu J, Niu Y: Quantitative prediction of MHC-II binding affinity using particle swarm optimization. Artificial intelligence in medicine 2010, 50:127–132.View ArticlePubMedGoogle Scholar
- Dimitrov I, Garnev P, Flower DR, Doytchinova I: EpiTOP-a proteochemometric tool for MHC class II binding prediction. Bioinformatics 2010, 26:2066–2068.View ArticlePubMedGoogle Scholar
- Nielsen M, Lund O, Buus S, Lundegaard C: MHC Class II epitope predictive algorithms. Immunology 2010, 130:319–328.View ArticlePubMedGoogle Scholar
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M: NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61:1–13.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S: NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2007, 2:e796.View ArticlePubMedGoogle Scholar
- Zhang H, Lund O, Nielsen M: The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics 2009, 25:1293–1299.View ArticlePubMedGoogle Scholar
- Jacob L, Vert JP: Efficient peptide-MHC-I binding prediction for alleles with few known binders. Bioinformatics 2008, 24:358–366.View ArticlePubMedGoogle Scholar
- Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O: Learning MHC I--peptide binding. Bioinformatics 2006, 22:e227–235.View ArticlePubMedGoogle Scholar
- Zaitlen N, Reyes-Gomez M, Heckerman D, Jojic N: Shift-invariant adaptive double threading: learning MHC II-peptide binding. J Comput Biol 2008, 15:927–942.View ArticlePubMedGoogle Scholar
- Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V: MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic acids research 2005, 33:W172–179.View ArticlePubMedGoogle Scholar
- Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V: Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC bioinformatics 2008,9(Suppl 12):S22.View ArticlePubMedGoogle Scholar
- Zhang H, Lundegaard C, Nielsen M: Pan-specific MHC class I predictors: a benchmark of HLA class I pan-specific prediction methods. Bioinformatics 2009, 25:83–89.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O: Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol 2008, 4:e1000107.View ArticlePubMedGoogle Scholar
- Justesen S, Harndahl M, Lamberth K, Nielsen LL, Buus S: Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immunome research 2009, 5:2.View ArticlePubMedGoogle Scholar
- Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B: The immune epitope database 2.0. Nucleic acids research 2010, 38:D854–862.View ArticlePubMedGoogle Scholar
- Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O: Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 2003, 12:1007–1017.View ArticlePubMedGoogle Scholar
Copyright
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.